水合电子还原降解水中PFAS 的研究进展
2024-03-08包一翔胡嘉敏钟金魁李井峰兰州交通大学环境与市政工程学院甘肃兰州730070北京低碳清洁能源研究院煤炭开采水资源保护与利用全国重点实验室北京1009甘肃省黄河水环境重点实验室甘肃兰州730070
杨 烨,包一翔,胡嘉敏,吴 敏,钟金魁,李井峰 (1.兰州交通大学环境与市政工程学院,甘肃 兰州 730070;.北京低碳清洁能源研究院煤炭开采水资源保护与利用全国重点实验室,北京 1009;3.甘肃省黄河水环境重点实验室,甘肃 兰州 730070)
全氟和多氟烷基化合物(PFAS)是一类碳原子上连接的H 原子被F原子全部或部分取代后形成的含有C−F 键的化合物[1-2].常见的PFAS 有全氟辛烷磺酸 (PFOS,CF3(CF2)7SO3H), 全 氟 辛 酸 (PFOA,CF3(CF2)6COOH) 与全氟己烷磺酸(PFHxS,CF3(CF2)5SO3H)等,结构由疏水疏油性的含氟碳链(CnHmF2n-m+1)与亲水官能团(如,磺酸基和羧酸基)组成(图1)[3-5].PFAS 中H 被F 取代后形成的C−F 键的键能较高(485.3kJ/mol)[6-7],且全氟结构保护了C−C键免受攻击[8],使得PFAS 具有热稳定性,化学稳定性,生物难降解性以及环境持久性,被称为“永久化学品”[9-10].
1951 年,美国3M 公司成功开发出PFAS,因其优异的化学稳定性,表面活性等性质,被广泛应用在工农业以及日常生活用品中[11-14].美国环境保护署(EPA)统计目前全球范围内分布着9000 多种不同的PFAS(包括其前驱体与代谢物)[15-16].PFAS 在不同环境介质(如大气,水体,土壤,生物等)中的积累和迁移能力不同,PFAS 的大气停留时间与其性质(包括挥发性,稳定性,分子量,链长和气−固相分配系数等)密切相关,最远迁移距离可达400km 以上[17].表层土壤中的天然有机质(NOM)可通过吸附或其他化学过程固定PFAS[18].此外,某些土壤矿物(如,铝硅酸盐,氢氧化铁,铝氧化物等)可作为PFAS 的吸附位点,从而抑制PFAS 的迁移.由于PFAS 具有较高的水溶性与较低的蒸汽压,使其更容易进入水环境[19-20],可通过江河,海洋等发生长距离的迁移[21-22].水体中PFAS浓度一般为几个ng/L,且在氟化工业园区,军事基地,飞机场以及化工业聚集地的水体中的浓度可高达几十至几千ng/L[23-25].
PFAS 易通过皮肤接触与饮食进入体内,与人体血液中的各种蛋白质有很强的结合能力,能够引起人体各个层次的毒性[26-28].近期有研究表明,血液中的PFAS可增加患重症新冠肺炎的风险[29],对人体健康造成了严重威胁.《关于持久性有机污染物的斯德哥尔摩公约》已经限制PFOA,PFHxS,PFOS 及其相关化学品的生产与使用[2,21,30].将PFAS 作为一个整体进行管控,已成为全球各国的共识[31-35].
如何高效去除水体中的PFAS 是当前亟待解决的难题.吸附,膜分离等相分离技术具有操作简单,去除率高等优点,已广泛应用于水中PFAS 的去除[36-38].但处理完成后的吸附剂与膜浓液的处置技术尚不成熟,常规的处置方法(填埋,焚烧等)易造成二次污染[39].因此与相转移技术相比,高效的降解技术才能更好地消除PFAS 的潜在风险.
由于PFAS 的结构稳定性,可行的降解技术十分有限.传统的生物降解对PFAS 几乎无效[40-44],热解技术对C−F 键破坏能力较差,存在能耗高,易造成二次污染等问题[45-48].高级氧化法(AOP)可通过光照,超声,电解等一系列手段产生高活性氧化性物种[49-50],攻击含 F 碳链与活性官能团的连接点位[40,45,51],破坏PFAS 的结构稳定性.但AOP 技术对PFAS 降解普遍效率不高,易产生大量的氟代降解产物,且无法降解稳定性更强的 PFOS 等磺酸类PFAS[43,52-55].高级还原技术(ARP)是近年来兴起的PFAS 降解技术,通常采用紫外激发光敏剂产生的强还原性水合电子(eaqˉ),实现PFAS 高效脱氟降解,极大降低了氟代中间产物的累积.F原子的电负性很高,更倾向于被电子攻击,进而破坏PFAS 中的C−F键,C−C 键实现降解[40,56-59].光生eaq‾技术中光敏剂(如SO32‾,I‾与Fe(CN)64‾等)易影响水体的化学稳定性,存在一定的环境风险.然而,该技术对于各类水体中常见的PFAS(包括PFOA 与PFOS 在内)降解较为彻底,同时F 矿化程度较高[59].因此,光生eaq‾仍是一种具有相当潜力的PFAS 降解技术,并且具备较好的工程应用前景[1,21,24].
本文综述了目前主要的光生eaq‾的产生原理及其对PFAS 的降解机理.阐述了水中的溶解氧,pH 值,共存物质,温度等关键水质参数对eaq‾产率和利用率的影响.讨论了PFAS 碳原子数,官能团种类,杂原子组成等对PFAS 降解机理的影响,以充分阐明eaq‾在PFAS 降解中的优势.通过对eaq‾研究发展历程及其对PFAS 降解技术的潜在问题进行全面综述,可为各类水中PFAS 污染控制提供技术参考.
1 水合电子概念与性质
1879 年,化学家Hogarth 等[60]首次在液氨中检测到溶剂化电子−氨化电子(1 个电子溶解在液氨中,氨分子对该电子发生了溶剂化作用),由此推测水在接受外部能量辐射后,可产生类似的溶剂化电子.1962年,Hart 等[61-62]根据吸收光谱波长700nm 左右短暂的特征吸收峰,正式定义了eaq‾的存在.
Kevan[63]通过电子自旋共振实验(ESR)阐释了eaq‾的理论结构模型,即数个水分子(一般为4,6,8 个)形成1 个空腔−“水化笼”.电子处在这样的“笼”中,以氢键与水分子结合.这种结构保证电子可在水体中单独存在(图1),但其在水中的存在寿命依然较短(10‾5s)[64].
但电子并不是完全地处在空腔中,仅40%的体积在空腔中占据空间,其余一部分与水化层中的配位水分子重叠,另一部分渗透入更远的氢键网络中(图2)[64].该模型兼顾了配位水化结构理论与氢键网络中电子的相互作用,因此eaq‾结构的核心问题不在于电子是否处在“水化笼”中,而是由3个不同部分共同组成了eaq‾的复杂结构.虽然eaq‾与常规的阴离子体系在水中的存在状态有相似之处,但是在扩散动力学上完全不同.Tay[65]通过分子动力学模拟,证明了电子的扩散是由溶剂的振动与平动动力学协同驱动,水分子之间构成的氢键网络增强了其迁移能力,使eaq‾比普通阴离子的扩散能力更强.

图2 电子部分处在氢键网络中部分与配位水分子重叠[64]Fig.2 Part of the electron resides in the hydrogen bond network and part overlaps with the coordinated water molecule
eaq‾作为亲核试剂,可与有机物发生氧化还原反应.当有机物分子含有如F, Cl, Br等电负性较高的卤素原子时,eaq‾可断裂有机物中的C−X 键并释放出卤素离子[66-67].因此,以eaq‾为主导的ARP 技术对PFAS的降解具有巨大潜力.
2 光生水合电子降解PFAS 的技术
水合电子通常由紫外光(UV)激发溶液中的光敏剂产生,目前亚硫酸盐与碘化物是研究较多的光敏剂.大量研究表明,紫外−亚硫酸盐法(UV/SO32‾)与紫外−碘化钾法(UV/KI)可有效降解水中的多种PFAS.
2.1 紫外−亚硫酸盐法(UV/SO32‾)
UV/SO32‾技术是一种通过UV 辐照SO32‾生成eaq‾和SO3‾•,实现PFAS 降解的技术.SO32‾光电离产生活性基团的链式反应如下[68-71](式1~4).
Song 等[59]以UV254(波长为254nm 的紫外光)构建UV/SO32‾体系降解水中的PFOA.反应24h 后PFOA几乎完全降解,脱氟率达88.5%以上.向反应体系中加入NO3‾与NO2‾消耗eaq‾后,PFOA的降解受到抑制,这证实eaq‾是导致PFOA 降解的关键物质.通过时间瞬态吸收光谱可以测定eaq‾在特定波长下的量子产率(特定波长的光下,1 个光量子生成eaq‾的量)[72-73],当把UV 的波长从258nm 缩短至194nm时,eaq‾的量子产率从 0.108mol/einstein 提高至0.391mol/einstein[74].因此,采用波长更短,能量更高的真空紫外光(VUV)可以有效提高eaq‾的量子产率.
Gu 等[66]对比了UV254与VUV185为2 种光源下,UV/SO32‾法对PFOS 的降解效果.反应完成后,PFOS降解率分别为85.8%和97.3%,VUV/SO32‾体系的降解速率比 UV/SO32‾体系快两倍以上.单独进行VUV185照射使PFOS 在4h降解了46.2%,而单独UV照射或仅加入亚硫酸盐对PFOS均无降解效果.原因是VUV 能使水分子电离,产生eaq‾与OH•等活性基团,协同降解溶液中的PFOS(式5~6)[75-76].因此,当UV/SO32‾体系以VUV 作为能量源时,可显著提升对PFAS 的降解效果.
在一定浓度范围内,提高SO32‾浓度可显著增强UV/SO32‾体系对PFAS的降解与脱氟效果.Song等[59]证实SO32‾浓度与eaq‾的量子产率是正相关的,将SO32‾浓度从0.5mmol/L 增加至20.0mmol/L 时,6h 内PFOA 的脱氟率从5.6%增至68.6%,符合伪一级动力学模型.Gu 等[66]将SO32‾浓度从1.0mmol/L 增加至5.0mmol/L 时,PFOS 的降解率从45.5%显著增加至87.1%,但将浓度进一步提升到 10mmol/L 与20mmol/L 时,降解率仅分别增加了6.0%与10.3%.原因是当体系中SO32‾浓度过高时,水中氧化性的S2O62‾积累,抑制eaq‾的产生,进而影响了对PFAS 的降解效果.
UV/SO32‾法光生eaq‾对除PFOA 与PFOS 外的其他PFAS 类物质也有较好的降解效果.Bao 等[77]研究了UV254下的UV/SO32‾还原体系与UV/S2O82‾氧化体系对六氟丙烯氧化物二聚酸(GenX)的降解效果.在UV/SO32‾体系中,反应2h 后GenX 被完全降解,6h 后脱氟率达到90%以上.而在UV/S2O82‾体系中,GenX 的降解率仅不到5%,与单独使用UV254照射降解趋势几乎相同.这说明,在UV/S2O82‾体系中,GenX 的降解的主要机理是UV 导致的直接光解,而OH•,SO4‾•等ROS 的氧化降解贡献度相对较小[52,78].因此,UV/SO32‾体系产生的eaq‾对PFAS 的脱氟降解更为高效.
PFAS中的端基Cl,醚键和C=C双键等反应活性位点,可以有效地增强UV/SO32‾体系对PFAS 的降解能力.随着国际上对PFOS的生产与使用的逐步限制,其替代品氯代多氟醚基磺酸(F-53B,图3)的生产与使用量大幅提高[79].Bao 等[58]探究了UV/SO32‾体系对镀铬废水中F-53B 的降解效果.反应30min 后,F-53B 的降解率达到90%以上,1h 后F-53B 彻底降解.F-53B 的结构为1 个醚键插入PFOS 的全氟碳链中,且末端的1 个F 被Cl 取代,改变了最低分子未占轨道(LUMO)组成,促进了eaq‾对F-53B 的降解[80-81].且SO32‾为电镀铬废水中含有的主要组分之一,因此UV/SO32‾法是一种降解复杂镀铬废水中F-53B 的可行技术.
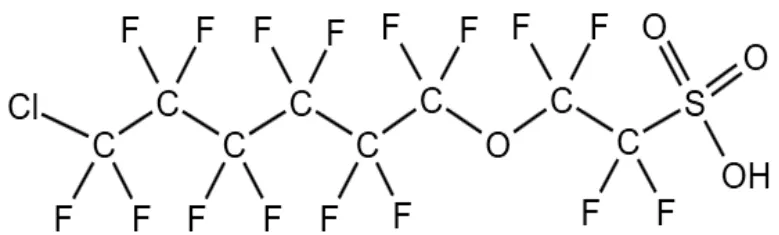
图3 F-53B 的化学结构[58]Fig.3 Chemical structure of F-53B
PFOS 的另一替代品−对全氟壬氧基苯磺酸(OBS,图4)是一种结构内同时含有C=C 双键,醚键,苯环与磺酸基团的PFAS.OBS 作为一种新型全氟表面活性剂,在中国年产量约3500t,常应用在水成膜泡沫灭火剂与化学强化采油剂中[82].Liu 等[83]探究UV/SO32‾体系对OBS 的降解效果,反应20min 后OBS完全降解,脱氟率为87.7%.eaq‾对OBS的降解并不会伴随碳链缩短,原因是OBS 支链上的C−C 键键能较高,难以直接断裂,而C=C 双键,醚键等结构为eaq‾提供了潜在的反应位点,易受到亲核攻击而断裂.在OBS 完全脱氟降解后,仍存在部分芳香类中间产物无法降解.原因是eaq‾对苯环结构几乎无降解效果,而UV/S2O82‾氧化体系产生的ROS 可以有效破坏苯环结构,具有更高的TOC 降解率与更快的降解速率(kOBS=1.05min‾1),但脱氟率较低(27.6%).因此在UV/SO32‾体系对OBS 脱氟完成后,可采用其他技术(AOP 法,生物降解等)进一步深度处理.

图4 OBS 的化学结构[83]Fig.4 Chemical structure of OBS
UV/SO32‾法对于还原降解PFAS 较为彻底,具有较高的氟回收率,这有助于减少PFAS 在环境中的积累和毒性影响.当采用VUV光源时,由于其具有更短的波长和更高的能量,可提高eaq‾的量子产率,并激发水电离产生活性物种,显著增强体系对PFAS 的降解效果.SO32‾的光分解产物为SO42‾与S2O62‾,S2O62‾可通过一系列反应转化为SO32‾再次参加反应.虽然SO42‾是自然水体中主要离子之一,在低浓度下不会造成环境风险,但在水处理工艺中加入SO32‾可能会破坏水体的化学稳定性,对工艺流程和排水管网产生不利影响[23,26,53].因此,应当对UV/SO32‾的应用场景进行合理定位,比如用于膜浓液处理等,此外,需要开发更加绿色高效的水合电子还原降解PFAS 的技术.UV/SO32‾技术目前处于应用基础研究阶段,针对复杂的PFAS 污染问题,仍需持续对UV/SO32‾降解反应机理进行研究,优化技术参数,减少次生风险.
2.2 紫外−碘化钾法(UV/KI)
I‾在吸收UV 能量后跃迁为激发态((I•,H2O‾*))(式7),该状态十分不稳定,可衰变回I‾与H2O(式8)或分解为一种笼状中间态产物((I•,e‾))(式9),该状态下电子游离在I 原子附近,最终解离产生I•与eaq‾(式10)[84-87].
I‾上的电子转移到溶剂(CTTS)形成I‾*CTTS激发态(式11),此即为电子脱离I‾的临界状态,此时游离在I 原子附近的电子被称为光分离电子(etrapped‾),易发生电子溶剂化反应.该过程中电子能级发生跃迁,首先变为最低激发态预溶剂化的湿电子(ewet‾),最终转变为基态平衡溶剂化的eaq‾(式12),3 种电子之间的跃迁极为迅速,时间尺度在飞秒量级之内.因此,由式12 可知UV/KI 体系中I•与eaq‾是成对产生的,I•在溶液中发生一系列反应(式13~17)[88].
Hori 等[78]利用时间瞬态吸收光谱方法发现,UV254下I‾能够生成eaq‾.Qu 等[89]采用UV/KI 体系对水中的PFOA 进行处理,6h 后,其降解率达到了93.3%,脱氟率为76.8%,14h 后,PFOA 脱氟率达到98.8%.经用该方法处理来自中国江苏省特氟龙制造厂实际PFAS 工业废水,主要成分为PFOA 与某些短链全氟羧酸(PFCA).经12h 处理后,废水中96%的PFOA 被降解,其他PFCA 的降解率在38.3%~62.0%之间,降解产物为 F‾与一些短链羧酸.这证实了UV/KI 法降解实际PFAS 工业废水的有效性.Park等[90]在开放环境下验证了UV/KI 体系可以有效降解包括PFOA, PFOS, PFHxS, 全氟己酸(PFHxA),全氟丁酸(PFBA),全氟丁烷磺酸(PFBS)在内的6 种PFAS.
通过增加初始KI的浓度可提升对PFAS的降解能力,但当浓度增加至10mmol/L以上时,UV/KI体系对PFAS 的降解将受到抑制.Qu 等[89]将体系中KI 浓度自0.1mmol/L 提高至0.3mmol/L 时,PFOA 的降解速率持续增加,峰值约是室温下直接光解速率的12倍.但当浓度进一步升高,PFOA 的降解速率显著降低.这可能是因为随着KI 初始浓度的增加与反应的进行,溶液中氧化性的I3‾累积(式15~17),将消耗参与还原降解eaq‾的量,抑制了PFAS 的降解(式18~19)[91].
UV/KI 体系中加入腐殖酸(HA)或黄腐酸(FA)等有机物可降低I3‾累积带来的负面效果.Sun 等[92]使用UV/KI+HA 体系降解水中的PFOS,反应1.5h 后,PFOS 的降解率与脱氟率分别为86.0%与55.6%;未加HA 的UV/KI 体系中,PFOS 降解率51.7%,脱氟率为4.4%.HA 使UV/KI 体系降解能力提高的原因是:(1)HA 自身吸收UV 能量,释放出eaq‾(式20);(2) HA易与I3‾反应生成含I 有机物(orgI),在UV 辐照下分解为I‾,形成了I‾生成eaq‾的良性循环,减少了eaq‾的消耗(式21~22);(3)HA 中的某些特殊结构,可作为电子载体,增强eaq‾的迁移能力,使其更易与PFAS 接触;(4)I‾和HA 可与PFAS 中某些官能团缔合,破坏原本稳定的化学结构.以上原因协同促进了PFOS 的降解.
研究发现,向UV/KI 体系中加入SO32‾可提高对PFAS 的降解能力[93].I 与S 衍生物间的协同作用,使降解体系的耗能大幅降低,同时提高了eaq‾的量子产率,但该体系增强PFAS 降解效果的关键机理仍不清楚[93].
综上所述,UV/KI 法降解PFAS 仍有很大的研究潜力与价值,但I‾在水处理过程中易产生包括碘乙酸,碘仿在内的多种碘代衍生物,其具有比氯代衍生物更强的遗传毒性与细胞毒性[85,92].因此,在未来UV/KI 法降解PFAS 技术的研究中,应当着重考虑该使用技术处理目标水的适用领域及KI 投加量的边界,并结合实际水质参数对处理过程中碘代衍生物的浓度水平变化进行监测.
2.3 其他可光生水合电子降解PFAS 的技术
除上述提到的UV/SO32‾与UV/KI 法外,可光生eaq‾降解PFAS 的技术还有紫外−亚铁氰化物法,紫外−部分有机物与等离子体等[94].
2.3.1 紫外−亚铁氰化物法(UV/Fe(CN)64‾) Huang等[95]以波长为266nm 的UV 激光(能量为3.0mJ/pulse)活化K4Fe(CN)6,在激光闪光光谱690nm 处,有eaq‾的特征吸收峰出现.N2O 可快速清除降解体系中的eaq‾(式23)[96],将其加入体系后eaq‾的特征吸收峰消失,证明是Fe(CN)64‾吸收UV 能量释放出eaq‾(式24),且Fe(CN)64‾的eaq‾产率高于I‾[74],说明UV/Fe(CN)64‾对PFAS的降解潜力高于UV/KI法.通过降解实验,证实 UV/Fe(CN)64‾法可有效降解水中的PFCA.
2.3.2 紫外−光敏性有机物 芳香类化合物(可分为具有供电子基团的苯环衍生物与具有五元环的杂环化合物等)在UV 照射下可电离生成eaq‾[97].Gu等[98-99]研究了UV/苯酚体系光生eaq‾的机理,加入一氯乙酸作为eaq‾的指示物(式25),通过时间瞬态吸收光谱测得量子产率约为0.250mol/einstein.光生eaq‾的机理是:苯酚吸收UV 能量转化为激发态,之后分解产生eaq‾与C6H5O•(式26~27).
Tian 等[100]采用低压汞灯浸没式发射UV254构建UV/吲哚+蒙脱石体系(式28).由于eaq‾容易被伴生吲哚自由基自淬灭,导致参与还原降解的eaq‾的量减少(式29).蒙脱石晶体片层带负电荷且具有空间限域效应,降解反应过程中吲哚自由基被吸附至蒙脱石层间的晶层固定,使其无法淬灭eaq‾.此外,蒙脱石层间由长碳链(C16)构成的有机相可以阻止氧化性物质进入,保护层间PFAS 的还原降解免受外部环境影响,提高了体系稳定性与降解能力.
Sun 等[101]研究了UV/硝基三乙酸(NTA)体系对PFOS 的降解能力.反应10h 后,PFOS 降解率与脱氟率分别达到了 85.4%与 46.8%.在相同条件下,UV/SO32‾体系对PFOS 的降解率与脱氟率为60.1%与26.9%,UV/NTA 法对PFOS 的降解速率高于UV/SO32‾法,遵循伪一级动力学方程.通过ESR 实验发现,NTA 中的羧基与胺基作为主要的反应位点,诱导水光解与离子化产生eaq‾与OH•等活性基团(式7~8).经此反应后,NTA 可消耗水中的OH•,避免了eaq‾在体系中的自淬灭.这导致可与PFAS 发生反应的eaq‾量增加,进而提升对PFAS 的降解效果.

表1 光生水合电子降解PFAS 的技术Table 1 Photo-generated hydrated electron degradation technologies of PFAS
2.3.3 等离子体法 通过激光诱导水分子电离可产生包括eaq‾在内的多种活性基团,形成具有高降解能力的等离子体−液体界面.该高能界面可降解水体中的多种PFAS 及其前体物质[102].等离子体系中复合的多重协同反应,可在短时间内破坏PFAS 中的全氟甲基结构(−CF3),同时切断碳链与官能团的连接[103].等离子体对长链PFAS 的降解不受水体中其他共存物质的影响,降解率可达到99%以上,在10min 内,PFOA 与PFOS 的降解率均达到90%以上[104].此过程在降解PFAS 时无需添加任何外部化学物质,不产生二次污染[105],但存在能耗高,对于短链PFAS 降解能力较差等问题.
综上所述,eaq‾还原降解PFAS 的技术具有反应速率快,降解较为彻底,F 矿化程度高等优点.然而,该技术需在碱性环境,无氧气氛,持续UV 照射下进行.此外,添加的高浓度光敏剂可能对水处理系统产生不利影响,这些因素都制约了它在实际工程中的应用.因此,寻找出一种反应条件简单,降解效率高,避免产生含光敏剂成分的副产物的 eaq‾还原降解PFAS 的技术将是未来研究的重点.同时还可将光生eaq‾还原技术与其他处理技术耦合,以实现对PFAS的高效处理.例如,利用电化学氧化或AOP 法整合该技术,也许能够更为彻底地降解PFAS,对其非氟化有机结构实现完全降解.这种综合应用的方法将有助于克服单一技术存在的局限性,从而最大程度的提高对PFAS 的降解能力.
3 光生水合电子降解PFAS 的机理
eaq‾对PFAS 的降解机理与全氟碳链结构,官能团种类,C−F 键性质(键长,键能),电子分布密切相关.尽管不同PFAS 的降解机理有所差异,但官能团相同(如羧基,磺酸基等)或碳链结构相似的PFAS,降解机理有共通之处.
3.1 全氟羧酸
PFCA 是一类具有代表性的PFAS,其结构由全氟碳链与羧基组成.Song 等[59],刘娇琴等[109]发现eaq‾还原降解PFOA 的中间产物包括C7F14HCOO‾,C7F13H2COO‾以及一些短链PFCA.降解产物种类不同的原因是,eaq‾对PFCA 的还原降解存在2 种机理:(1)C−F 键断裂发生H/F 置换;(2)经DHEH 过程断裂C−C 键,生成短链PFCA.
3.1.1 H/F 置换 eaq‾降解PFCA 的第1 种降解机理为H/F 置换(图5)[110].当PFCA 接收到第1 个eaq‾时,通过模拟几何优化发现α-C 上的C−F键有明显的拉伸,最先断裂进而发生H/F置换,主要原因是该位置上C−F 键的解离能(BDE)最低.Bentel 等[110]经密度泛函理论(DFT)计算了部分PFAS 中的C−F键的BDE.结果显示,位于PFAS 碳链末端的−CF3中的“一级C−F 键”的BDE 高于位于碳链中部的−CF2−上的“二级C−F 键”.随着PFAS 全氟碳链长度增加,其一级与二级C−F 键的BDE 逐渐降低(图5).位于碳链中部−CF2−上的C−F键BDE更低,并且与eaq‾有最强的亲和力,具有最高的反应活性,最易发生断裂.因此, eaq‾对长链PFCA 的还原降解效果优于短链PFCA,降解中间产物的浓度均呈现先增后减的趋势[87].

图5 PFCA 降解与脱氟机理及PFAS 中C−F 键的BDE (kcal/mol)[110]Fig.5 Degradation and defluorination mechanism for PFCA and BDE of C−F bonds in PFAS (kcal/mol)
Qu 等[89]在eaq‾对PFOA 的降解过程中检测到α-C 上至多两次H/F 置换形成的降解中间产物(Cn-1F2n-1CH2COO‾,式30~34),提出如果PFCA 的碳链足够长(碳原子数≥5),那么在除α-C 以外的其他C原子上也有可能发生H/F 置换.Cn-1F2n-1CH2COO‾经UV 诱发分解为Cn-1F2n-1•,COO‾•与甲烯基(:CH2)(式35),Cn-1F2n-1•与COO‾•重组形成Cn-1F2n-1COO‾(式36),该产物较母体PFCA 脱去1 分子−CF2−后,进入下一反应周期.然而,H/F置换无法实现PFCA的完全降解,原因是随着反应进行,Cn-1F2n-1•的浓度降低,导致Cn-1F2n-1•与羧基的复合变得困难.
3.1.2 DHEH 过程 Hori 等[78,111]在重氧水中光生eaq‾ 降解 PFOA, 检测到[C6F13C(16O)(16O)]‾,[C6F13C(16O)(18O)]‾与[C6F13C(18O)(18O)]‾3 种降解产物.说明PFCA 的降解过程有多种C−C 键断裂的方式.另一个PFCA 降解机理是DHEH 过程(涵盖了脱羧(式37~38),羟基化(式39),脱氟(式40)与水解(式41)四个步骤).在UV 与eaq‾的协同作用下,碳链与官能团间的C−C 键断裂,生成CnF2n+1•与COO‾•,CnF2n+1•,之后经羟基化与水解过程,转化为相比母体PFCA脱去1 个CF2单元的短链PFCA.PFCA 经DHEH 过程不断缩短碳链[112-113],直至完全降解,产物主要为CO2, H2O 与F‾[114-115].
DHEH 过程与H/F 置换是平行存在的,互为竞争性机理.在H/F 置换后,形成的Cn-1F2n-1-CH2COO‾中的−CH2−基团,增强了全氟碳链与羧基间C−C 键的强度,增大了脱羧难度,导致DHEH 过程难以启动[113].
除了H/F置换与DHEH 过程外,PFCA 的降解过程还涉及许多尚未明确的复杂机理,如Ren 等[106]在UV/SO32‾体系对PFOA 的降解产物中,检测到羧基α-C 上存在磺酸基团的降解产物(C7F14SO3HCOO‾),推测是SO3‾•可发生类似H/F置换的SO3‾•/F置换(式42~45,图6),脱羧后转变为短链全氟磺酸(PFSA),增大了降解难度.

图6 UV/SO32‾降解体系中发生类似于H/F 转换的SO3‾•/F置换[106]Fig.6 SO3‾•/F exchange occurs in the UV/SO32‾ degradation system similar to H/F exchange
3.2 全氟磺酸
PFSA 由全氟碳链与磺酸基团(−SO3‾)构成.eaq‾降解PFSA 的中间产物主要为短链PFSA 与PFCA,而不同的中间产物代表着特定的降解机理.PFSA 的降解主要有3 种机理:脱硫,H/F 置换以及全氟碳链中部的C−C 键断裂[114,116].
3.2.1 脱硫 在脱硫过程中,eaq‾主要攻击全氟碳链与磺酸基团连接的C−S 键,模拟几何优化结构中C−S 键的伸长验证了这一理论.PFSA 中C−S 键的键能(272kJ/mol)显著低于C−C 键的键能(346kJ/mol),且C−S 键长大于C−C 键,相对更容易断裂[117].
PFSA 通过2 种脱硫路径,最终转化为PFCA.第1 种是C−S 键直接断裂,生成CnF2n+1‾与SO3‾•(式46~47),CnF2n+1‾羟基化为CnF2n+1OH(式48),最终转变为 PFCA(式 43~44).另 1 种是生成 CnF2n+1‾•与SO32‾(式49),CnF2n+1•经一系列反应(式39~41)转变为PFCA[110].但eaq‾的还原降解体系通常为碱性,因此第2 种脱硫路径存在的可能性更高[110].
3.2.2 H/F 置换 PFSA 的第2 种降解路径是H/F置换.PFSA 的“二级C−F 键”具有较低的BDE(图3),因此H/F 置换通常是连续地发生在全氟碳链中部的碳原子上(式50~52).然而,尽管磺酸基团β-C 上C−F键的BDE 同样较低,却无法发生H/F 置换,这表示PFSA 内单个C−F键的BDE不是决定降解机理的唯一因素[90,92].
3.2.3 C−C 键断裂 PFSA 的第3 种降解机理是C−C 键直接断裂.全氟碳链中间位置的C 原子处在PFSA 的LUMO 上,这些−CF2−在吸收eaq‾后将削弱与其他碳原子之间的联系.C−C 键的断裂通常有2种方式:(1)−CF2−吸收eaq‾后带有负电荷,易与水中的H+结合,发生电子转移引发C−C 键的断裂[87].但eaq‾的降解体系大多为碱性,因此实现H+与PFSA 的结合难度较大;(2)PFSA 吸收eaq‾后直接断裂C−C 键,产生短链全氟烷基阴离子与含磺酸基的全氟烯烃,之后转化为PFCA 与短链PFSA,最终完全降解.Gu等[66]以 UV/SO32‾法降解 PFOS,检测到 H/F 置换,C−C 键断裂的降解中间产物与一些短链PFCA,证实在PFSA 的还原降解过程中3 种降解机理协同进行,共同促进PFSA 的降解.
3.3 氟调聚物
氟调聚物是一类具有在全氟碳链的一端与非氟化的烷基或官能团相连结构的PFAS.
3.3.1 氟调聚羧酸 氟调聚羧酸(FTCA)是PFCA的前体物质,其结构特征是在全氟碳链与羧基间连接着数个−CH2−基团,这显著提高了FTCA的稳定性,难降解性以及含氟碳链对官能团的束缚能力.eaq‾对FTCA 和PFSA 的脱氟速率相近,且6:2FTCA 阴离子自由基中的“二级C−F 键”同样出现明显的拉伸[110].这些结果证实了eaq‾对FTCA 与PFSA 降解机理具有相似性,两者的降解过程都开始于“二级C−F 键”上的H/F 置换,之后经C−C 键断裂等一系列反应转化为PFCA.
3.3.2 氟调聚磺酸 氟调聚磺酸(FTSA)与FTCA的结构相似,可作为PFOS 与PFOA 的有效替代品.Bao[108]提出6:2FTSA 的降解可从全氟碳链的任意碳原子上开始,C3 与C4 合计的LUMO 贡献率超过58%,是最易受eaq‾攻击的反应位点.此外,全氟碳链越长,FTSA 的降解速率越快.这与Bental[110]阐述PFAS 碳链长度与碳链中部C−F键的BDE 成反比在实验结果上保持一致.
3.4 其他PFAS
3.4.1 六氟环氧丙烷寡聚酸 全氟醚羧酸(PFECA)与全氟醚磺酸(PFESA)因其优秀的性质广泛应用于工业生产中,其结构特征是在PFCA 或PFSA 中引入1 个或多个醚键.六氟环氧丙烷(HFPO)寡聚酸是PFECA 中一类重要的PFOA 替代品[118].Bao 等[21]从分子结构的角度提出,eaq‾对HFPO 寡聚酸的降解与LUMO,BDE 与DhAs(二面角,常指分子中由四个原子组成的2 个平面之间的夹角,用于描述分子的扭转角度)密切相关.在HFPO 寡聚酸结构中,醚键与羧基显著改变了分子的DhAs.HFPO-DA的主链是线性的,而随着醚键数的增加,HFPO-TA与HFPO-TeA 的主链逐渐弯曲(图7),使其更容易接触到体系内的eaq‾,并削弱了−CF3基团对分子结构的保护作用.然而,这种分子构型变化对于BDE无影响[21].
图7 HFPO-DA,HFPO-TA 和HFPO-TeA 的分子构型[21]Fig.7 Molecular configurations of HFPO-DA, HFPO-TA and HFPO-TeA
根据LUMO 分布得知,eaq‾几乎可以在HFPO 寡聚酸除羧基外的所有位点上发生反应,而醚键尤其容易受到攻击.HFPO-TA 和HFPO-TeA 通过一系列化学反应过程,包括烷基化,醇化,酰氟化与羧基化(简称“4A 过程”),断裂醚键并生成HFPO-DA,全氟丙酸(PFA),全氟乙酸(TFA)等降解产物.根据福井函数对GenX 的(HFPO-DA 铵盐)LUMO 分析,f+等势面(易受亲核攻击的部分)在分子的非离子端(图8)[119],而结构中C−O 键BDE 最低,因此GenX 的降解也始于醚键断裂,后经“4A 过程”最终完全降解.GenX 与PFCA 的降解突出了醚键的重要性,PFCA依赖H/F 置换与DHEH 过程脱羧,随后逐步缩短碳链,而“4A 过程”始于醚键的断裂,等同在PFCA 的全氟碳链中多引入了1 个活性反应位点,从而加速了GenX 的降解.
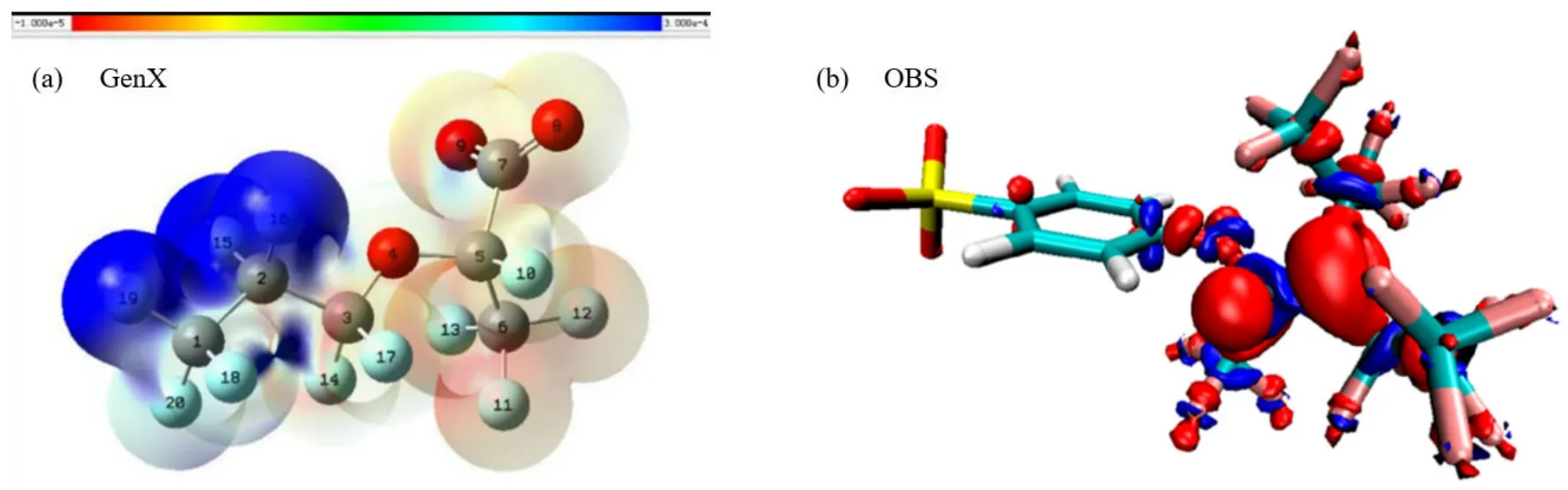
图8 GenX[77]与OBS[83]的LUMO 等势面Fig.8 The LUMO contour surface of GenX and OBS
3.4.2 OBS OBS 结构内同时存在醚键,C=C 双键与磺酸基团.f+等势面在醚键,C=C 双键以及全氟末端的碳原子上,其中C=C 双键的f+最高.然而,由于PFAS 中F 的高电负性,eaq‾更倾向于攻击C−F 键而非C=C 双键,且C=C 双键的α-C 具有除C=C 双键外最高的f+值,因此成为了最易发生H/F置换的位点(图8).在首次H/F 置换后,H 与β-C 上的F 一同脱出,形成新的C=C 双键,削弱了H 对H/F 置换的抑制作用.另一机理为醚键断裂生成全氟烷基自由基,发生羟基化或与磺酸基团结合后,在C=C 双键的2 个α-C 上连续地发生H/F 置换,形成2 个新的C=C 双键,C=C 双键极不稳定,最终在eaq‾的作用下完全降解为非氟化有机物与F‾.
3.4.3 F-53B F-53B 结构内含有醚键,磺酸基团与端基Cl(图3).脱氯与醚键断裂为F-53B 还原降解的2 条路径(图9).
在第1 条路径中,F-53B 的碳链末端发生H/Cl置换,随后断裂醚键,生成全氟烷基自由基与含氟磺酸阴离子自由基.第2 条路径跳过了H/Cl 置换过程,直接断裂醚键,产生与路径1 相似的降解产物(但Cl未被H 取代).两种路径的降解产物在eaq‾作用下,通过H/F 置换进一步降解,最终实现F-53B 的完全脱卤,但路径2 的解速率较低.
Bao等[58]计算了PFOS,F-53 与F-53B 的垂直电子亲合能(VEA),衡量1 个中性分子或原子在其基态下吸附1 个电子形成阴离子时的能量变化,用以说明对降解的影响.PFOS 的VEA(109.1kJ/mol)高于F-53(85.3kJ/mol)低于F-53B(132.2kJ/mol),这与降解速率的大小保持一致,即 F-53B>PFOS>F-53.当PFOS 的全氟碳链中引入氧原子时,VEA 反而会降低,但当全氟碳链末端F被Cl 取代时,VEA 显著提高(即使结构中存在O).经过DFT 计算,醚键的插入与碳链末端Cl 的替换,导致F-53 与F-53B 的LUMO等势面发生显著变化(图9).在F-53 与PFOS 中,碳链末端的F 对LUMO 的贡献率为0,Cl 对F-53B 的LUMO 贡献率为21.58%,碳链末端C 原子对F-53B的LUMO 贡献率为30.61%,远高于F-53(10.57%)与PFOS(5.41%),且在F-53B 中C−Cl 键的BDE 低于C−F 键[58].因此,端基Cl 是促进eaq‾对F-53B 还原降解的主要原因.
综上所述,eaq‾对PFAS 的降解主要通过H/F 置换,DHEH 过程以及“4A 过程”等途径进行.其中,结构中的醚键,C=C 双键与端基Cl 等可以改变PFAS 的分子构型与LUMO 分布,成为或使其α-C 转变为易受亲核攻击的反应位点.虽然带有不同官能团的PFAS具有不同的降解机理,但它们的降解活化能无显著差异,这表明eaq‾对PFAS的还原降解是一种扩散控制过程[69,119-120].因此,确保eaq‾可以充分接触目标PFAS(如在带电材料内部反应或是清除体系内的氧化性物质等)可以有效提升eaq‾对PFAS 的降解能力.
4 影响光生水合电子降解PFAS 的因素
eaq‾在合适的条件下可高效地降解水中的PFAS,但体系初始pH 值,溶解氧含量与温度,共存物质等因素都可能通过不同的机理影响eaq‾对PFAS 的还原降解.
4.1 初始pH 值
初始pH 值可显著影响eaq‾对PFAS 的降解效果[45,121].Song 等[59]研究了UV/SO32‾体系初始pH值对PFOA 降解的影响.当pH 值小于6 时,UV/SO32‾体系几乎无法降解水中的PFOA,但当pH 值持续升高至8 以上时,PFOA 的脱氟率显著提升(≥50%),pH 值升高至10.3 时达到最大值.这一结果表明,在碱性环境下,UV/SO32‾体系对PFOA 的降解效果更为显著.
韩慧丽等[76]探究了VUV/SO32‾体系初始pH 值对PFOS 降解的影响,当pH 值从6 提高至10 时,PFOS 降解率从60.4%提高至97.3%,脱氟率从9.0%提高至63.8%.出现这一现象的原因是:在pH 值较低的环境下,H+会消耗水中的eaq‾(式53~55),每当pH值上升1 时,H+与eaq‾反应的速率常数就会提升1 个数量级,在降解体系中微小的pH 值变化都会对PFAS 的降解产生重大影响[35,122].并且pH 值的大小决定了S 在水中的存在形式,当pH 值大于9.2 时,99%以上的S以SO32‾的形式存在[53],提高了光生eaq‾的量子产率.
在VUV辐照下,水电离产生OH•与SO32-反应生成SO3‾•与OH‾(式56)[76],导致随着反应的进行,体系pH值缓慢升高.Qu等[123]指出改变初始pH值不会对PFAS 降解机理造成影响,但pH 值的升高会促进该反应的进行,加快降解速率.
4.2 溶解氧含量与温度
Song 等[59]对比经过N2曝气30min 与开放环境下,UV/SO32‾体系对PFOA 的降解效果.在无氧气氛下,PFOA 的24h 脱氟率达到了88.5%,而开放环境下PFOA 脱氟率仅为6.4%,证实降解体系中的溶解氧(DO)会对PFAS 的脱氟降解产生显著的抑制作用.主要原因是O2对eaq‾的淬灭作用(式57~58)[123],导致体系内eaq‾的量减少.Gu 等[107]使用高压汞灯作为光源构建UV/SO32‾体系降解溶液中的PFOS,反应开始前未对水中的DO 进行处理,反应仅30min 后,PFOS降解率几乎达到100%.高压汞灯可发射更宽的波长跨度的UV 与更高密度的光子,使降解体系中eaq‾产率大幅上升,抵消了DO 的淬灭作用.因此,在eaq‾对PFAS 的降解过程中应尽量避免或抵消因DO 造成的影响.
在光生eaq‾体系中,PFAS 的降解率与脱氟率随着温度升高而增加,原因是eaq‾同样遵守分子热运动规律,温度升高会增加eaq‾与PFAS 碰撞的概率.此外,水中的DO 溶解度会随着温度的上升逐渐降低,从而促进PFAS 的降解.但当超过临界温度后,继续升高温度反而会抑制PFAS 的降解.原因是当温度过高时,体系内会发生伴生ROS 对eaq‾的自淬灭现象,导致参与还原降解的eaq‾的量减少[106].因此,需要将光生eaq‾体系控制在适当的温度内操作,以获得最佳的PFAS 降解效果.
4.3 共存物质
4.3.1 共存阴离子 在eaq‾降解PFAS 的体系中,共存物质会对降解效果产生不同程度的影响.
Cl‾对于PFAS 的降解无显著影响,即使在含有极高浓度的Cl‾水体中,PFAS 的降解率与脱氟率也几乎不发生变化[106].
PO43‾对PFAS 的降解与脱氟具有轻微的抑制作用.有研究表明,向UV/SO32‾体系中加入少量PO43‾,PFOA 的24h 脱氟率由90%下降至82%[106].由于PO43‾无法吸收UV254,这种轻微抑制作用的根本原因是PO43‾对eaq‾的淬灭作用.尽管PO43‾的存在对PFAS 的降解影响不大,但是仍应尽量避免向体系中引入PO43‾.
NO3‾作为eaq‾的淬灭剂,可以迅速的消耗eaq‾,对PFAS 的降解有显著的抑制作用[42].当降解体系中存在1mmol/L 的NO3‾时,PFAS 在2h 内的降解受到抑制[41],主要原因是eaq‾与NO3‾反应的优先级高于PFAS,当NO3‾被完全消耗后才开始PFAS 的降解.
低浓度CO32‾对PFAS 的降解无显著影响,但当体系中CO32‾浓度过高时会发生UV 阻断效应,导致光敏剂无法吸收到足够的能量解离出eaq‾,且高浓度的CO32‾会消耗体系中的eaq‾.以上2 种因素协同作用下导致参与还原降解的eaq‾的量降低,表现出对PFAS 降解的显著抑制作用[125].
4.3.2 可溶性有机物 可溶性有机物(DOM)对PFAS 降解的影响与其浓度密切相关.在低浓度下,DOM 能促进PFAS 的降解,原因是部分DOM(如上文提到的HA)可与水中存在的Fe3+,I3‾等离子发生配位反应,避免了氧化性物质淬灭eaq‾.此反应形成的配合物还可充当电子载体,加强电子的扩散能力,提高对PFAS 的降解能力[126].
但体系中高浓度的DOM 具有强大的夺电子能力,将淬灭体系中的eaq‾.此外,高浓度DOM还会引起UV 阻断效应,使光敏剂无法吸收足够的UV 能量产生eaq‾[100],并伴生有机ROS,进一步抑制eaq‾对PFAS的降解[127].
5 结论与展望
PFAS 降解的关键在于实现分子中氟取代结构充分矿化以避免产生持久性的氟代降解产物.以eaq‾为主要活性物质的高级还原技术比以OH•,SO4‾•为主要活性物质的高级氧化技术在脱氟方面有更大优势.未来可在以下方面加强研究,以提高相关技术的工程应用可行性.
5.1 当前高级还原技术主要采用紫外光作为光源,亚硫酸盐等作为光敏剂,产生eaq‾,紫外光在透光率不高,水质复杂的水体中利用率会降低,不利于eaq‾生成,需要开发适应性更强的高级还原技术体系,实现不同水体中PFAS 的污染控制.
5.2 亚硫酸盐,碘化物,吲哚衍生物等作为光敏剂不宜应用于饮用水处理等领域,同时可能带来药剂投加后产生的处理问题甚至次生风险.可开发新型光生eaq‾还原技术体系,以提高不同应用场景下的技术可行性.
5.3 开发耦合技术体系,如高级还原技术耦合膜分离技术,活性炭和树脂吸附技术,对膜浓液,吸附饱和吸附剂进行处理/再生,同时可拓宽高级还原技术应用途径,提高PFAS 污染控制能力.
