Research Progress on Targets and Selective Inhibitors of Polo-like Kinase-1 (PLK-1)
2024-03-07XinWANGQinZENGGuangyingDU
Xin WANG, Qin ZENG, Guangying DU
University Collaborative Innovation Center of Shandong Province, Key Laboratory of Molecular Pharmacology and Drug Evaluation of Ministry of Education, Yantai University, Yantai 264005, China
Abstract In this paper, the biological function of PLK-1, the correlation between PLK-1 and tumors, and the latest research progress on PLK-1 inhibitors under study are reviewed, in order to provide references for the research and development of PLK-1 inhibitors.
Key words Polo-like kinase-1, PLK-1 inhibitor, Cell cycle, Mitosis, Cancer
1 Introduction
Cancer is a major public health issue worldwide. In China and developed countries, cancer is the main cause of death. It was estimated that approximately 4.82 million people in China and 2.37 million people in the United States were newly diagnosed with cancer in 2022, and 3.21 million and 0.64 million people were dead from cancer, respectively[1]. With the advancement of medicine and biology, the treatment methods for cancer have become diversified, such as surgical resection, radiotherapy, chemotherapy, targeted therapy, and immunotherapy, which have improved the clinical treatment effect to a certain extent. However, most patients often experience drug resistance/recurrence, and some patients have no response after treatment. Therefore, there is an urgent need to develop new cancer treatment methods or drugs to further improve the clinical treatment effectiveness of cancer patients.
Polo-kinases (PLKs) are a class of highly conserved serine/threonine protein kinases, and are widely distributed in eukaryotic cells. The family has discovered five subtypes of PLK-1 to PLK-5, among which PLK-1 is closely related to tumor occurrence and development. PLK-1 controls cell entry into mitosis by mediating specific substrate phosphorylation and is an important regulatory factor in the G2-M phase of the cell cycle. PLK-1 is also involved in centrosome maturation, spindle assembly, sisters chromatid cohesion, cytokinesis, DNA damage repair, autophagy, EMT and other important biological processes.
The expression of PLK-1 is up-regulated in a variety of tumor tissues, including gastric cancer, breast cancer, colorectal cancer, prostate cancer,etc., and the overexpression of PLK-1 is closely related to poor prognosis. Therefore, PLK-1 is a very promising anti-tumor drug target, and multiple PLK-1 inhibitors have entered clinical trials. The fastest progressing is Onvansertib from Cardiff Oncology, which is currently conducting a phase II clinical trial in combination with FOLFIRI/bevacizumab for first-line treatment of RAS mutant colon cancer (NCT06106308). PLK-1 inhibitors can exert anti-tumor effects through various mechanisms, including blocking the mitotic cycle of tumor cells, forming synthetic lethality with RAS mutation, enhancing the anti-tumor effects of chemotherapy drugs and anti angiogenic drugs. Therefore, the use of PLK-1 inhibitors alone or in combination with chemotherapy/anti angiogenic drugs is expected to become an important treatment method for KRAS mutant tumors.
In this paper, the structure and biological function of PLK-1, the correlation between PLK-1 and tumors, the action mechanism of PLK-1 inhibitors, and the latest research progress on PLK-1 inhibitors under study are mainly reviewed, in order to provide reference for the research and development of PLK-1 inhibitors.
2 PLK-1 structure
PLKs are a class of serine/threonine protein kinases widely distributed in eukaryotes, with five subtypes, among which PLK-1 to PLK-4 have similar structures. The amino end (N-terminal) is a highly conserved kinase domain (KD), and the carboxyl end (C-terminal) is a characteristic polo-box domain (PBD). The structure of PLK-5 is short, and it does not participate in cell cycle regulation.
PLK-1 has a T-ring at the N-terminal and two phosphorylation sites: serine at position 137 (S137) and threonine at position 210 (T210). S137 phosphorylation mainly plays a role in the S phase, while T210 phosphorylation mainly plays a role in mitosis. These two phosphorylation sites are directly related to PLK-1 kinase activity. The C-terminal contains two conservative polo boxes: PBD1 and PBD2 (Fig.1). PLK-1 binds to phosphopeptide epitopes of different substrates through PBD and is recruited to specific subcellular locations (such as centrosomes, centrosomes, spindles,etc.), then releases kinase domains to perform different biological functions. Normally, the PBD of PLK-1 binds to KD, inhibiting N-terminal phosphorylation and the activation of PLK-1. Therefore, PBD and KD of PLK-1 can both cooperate and antagonize each other. PBD’s recognition of substrates not only determines the intracellular dynamic localization of PLK-1, but also relieves its self inhibitory effect on the N-terminal kinase domain.
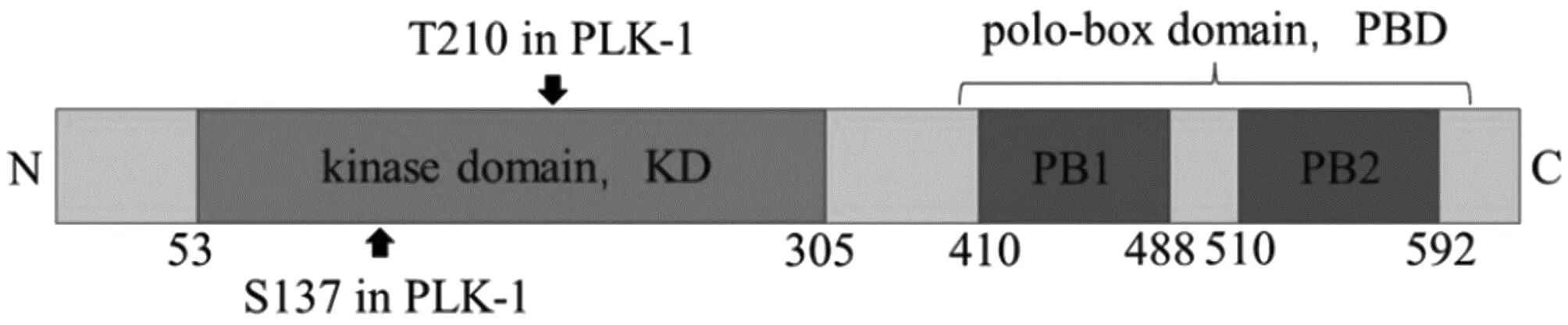
Fig.1 Primary structure of PLK-1
3 PLK-1 mediated signaling pathways
3.1SKA3-PLK-1-AKTsignalingpathwayIn laryngeal squamous cell carcinoma, PLK-1 mediates cancer progression through the SKA3-PLK-1-AKT signaling pathway[2]. Spindle and centromere related complex subunit 3 (SKA3) is involved in the occurrence and development of various tumors. In laryngeal squamous cell carcinoma, SKA3 regulates the activity of PLK-1 at the upstream, which in turn phosphorylates AKT. AKT is a threonine/serine protein kinase that plays an important role in tumor growth and differentiation[3]. The activated SKA3-PLK-1-AKT signaling pathway enhances glycolysis, provides energy for tumor cells, and promotes tumor proliferation, invasion, metastasis, and chemotherapy resistance.
3.2STAT3-PLK-1-AKTsignalingpathwayIn esophageal squamous cell carcinoma, PLK-1 mediates cancer progression through the STAT3-PLK-1-AKT signaling pathway[4]. Signal transduction and transcription activating factor 3 (STAT3) is abnormally activated in esophageal squamous cell carcinoma and is associated with regulating proliferation, survival, invasion, angiogenesis, and immune evasion of tumor cell. STAT3 activates PLK-1 and positively regulates its expression, which in turn phosphorylates AKT, forming the STAT3-PLK-1-AKT signaling pathway that promotes the progression of esophageal squamous cell carcinoma.
3.3STK39-PLK-1-CRAF-ERKsignalingpathwayIn hepatocellular carcinoma, PLK-1 mediates the progression of hepatocellular carcinoma through the STK39-PLK-1-CRAF-ERK signaling pathway[5-6]. Serine/threonine kinase 39 (STK39) interacts with PLK-1 and regulates its phosphorylation. PLK-1 induces epithelial-mesenchymal transition by activating the CRAF-ERK signaling pathway, promoting the progression of hepatocellular carcinoma.
3.4FOXM1andPLK-1signalingpathwayIn diffuse large B-cell lymphoma and esophageal cancer, the FOXM1-PLK-1 pathway is generally upregulated[7]. Forkhead box protein M1 (FOXM1) is highly expressed in various cancers and is involved in cell cycle regulation, cell proliferation, self-renewal, DNA repair, tumorigenesis, and drug resistance. FOXM1 and PLK-1 are mutually activated, thereby promoting the expression of cyclin CDK1 and cyclinB, and tumor cell proliferation.
4 Biological functions of PLK-1
4.1ParticipatinginmitosisPLK-1 is an important kinase that regulates the cell cycle and plays a crucial role in various stages of mitosis, including controlling the progression of mitosis, centrosome maturation and separation, spindle assembly, and cytoplasmic division and shedding[8]. In the G1phase of the cell cycle, various proteins aggregate at the starting point of DNA replication to prepare for replication. Cyclin E and cyclin dependent kinase 2 complex (cyclinE/CDK2) activates DNA helicase, triggering replication initiation and promoting cell entry into the S phase. The expression of PLK-1 first occurs in the G2phase, where Aurora A phosphorylates PLK-1, which is then recruited to the centrosome. Subsequently, PLK-1 phosphorylates cell division cycle 25C (CDC25C), while pCDC25C dephosphorylates and activates cyclin dependent kinase 1 (CDK1); PLK-1 phosphorylates WEE1, thereby phosphorylating and activating cyclinB. CDK1/cyclinB complex can promote the transition from G2phase to M phase (Fig.2). Therefore, the expression of PLK-1 is almost undetectable in the G1and S phases, and gradually increases in the G2phase and reaches its peak in the M phase. Dysregulation of PLK-1 activity can lead to mitotic defects, chromosome instability, and the development of tumors.
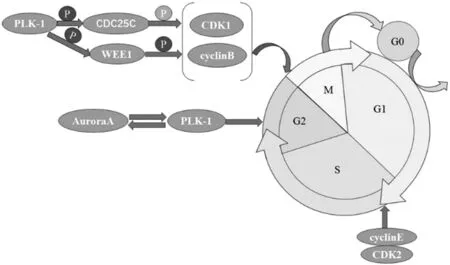
Fig.2 Molecular mechanism of PLK-1 involvement in cell cycle regulation
4.2RoleofPLK-1inDNAreplicationandDNAdamageresponseLong-term inhibition of the function of PLK-1 can lead to slow progression in the S phase, and studies have shown that PLK-1 is involved in DNA replication. The pre RC complex (pre-RC) is assembled in an orderly manner at the replication starting point, which is the binding of the multi protein origin recognition complex (ORC). ORC further recruits cell division cycle 6 (CDC6) and chromatin permissive and DNA replication factor 1 (CDT1) components, and enables small chromosome maintenance complex 2-7 (MCM2-7) to participate in DNA replication in the S phase[9]. Research has found that PLK-1 can interact and phosphorylate with several components of pre-RC, including ORC2, MCM2-7, and human histone acetyltransferase that binds to ORC1 protein (HBO1). If PLK-1 is missing, the formation of pre-RC is disrupted during the G1-S phase transition, leading to impaired DNA synthesis. Therefore, inhibiting PLK-1 in tumor cells can induce DNA damage, leading to tumor cell death. In the process of DNA replication, the activity of DNA topoisomerase is also crucial, as it helps to prevent DNA from becoming tangled during unwinding. Research has found that PLK-1 can phosphorylate DNA topoisomerase II α, and in turn affects DNA replication. The above data indicates that PLK-1 is necessary for DNA replication.
There is a certain probability of DNA damage during the process of DNA replication. If not repaired or improperly repaired, it may lead to mutations and ultimately form tumors. However, the body has a spontaneous safety mechanism called DNA damage response (DDR): after DNA damage, DNA damage checkpoints can be activated, cell cycle processes can be stopped, and DNA repair can be promoted.
The DNA damage checkpoint is divided into G1-S phase checkpoint, G2-M phase checkpoint, and spindle assembly checkpoint (SAC). When DNA damage occurs during the interphase of mitosis, ATM/CHK kinase is activated, inhibiting the activity of PLK-1. When DNA damage occurs during mitosis, G2-M phase checkpoint is activated, interfering the interaction between Aurora A and Bora/PLK-1 complex, and inhibiting the activation of PLK-1 mediated by Aurora A[10]. In addition, DNA damage can lead to the accumulation of inactive lysine monomethylation at position 209 of PLK-1, which can further inhibit the activity of PLK-1[11]. Premature activation of PLK-1 during DDR may cover DNA damage checkpoints, allowing cells to continue their cell cycle and potentially leading to genomic instability, resulting in the development of tumors.
4.3RoleofPLK-1inautophagyAutophagy is an adaptive mechanism that maintains cellular homeostasis, ensuring optimal redistribution of energy sources during cellular stress. Autophagy is a double-edged sword in tumors. On the one hand, autophagy has the characteristic of inhibiting tumors, and on the other hand, it can also promote tumor growth[12]. Mammalian rapamycin target (mTOR) is a key regulatory molecule for cell growth, playing an important role in balancing cell growth and stress-induced autophagy. mTOR forms mTOR complex 1 (mTORC1) together with mTOR related proteins and inhibits autophagy under normal conditions[13]. Studies have shown that PLK-1 can participate in the regulation of autophagy through its interaction with mTOR. PLK-1 expression is elevated in human glioma cells. Knocking down PLK-1 can enhance mTORC1 activity, inhibit autophagy, and induce caspase dependent apoptosis. Studies have found that it can enhance tumor cell sensitivity to radiation by inhibiting autophagy in tumors[14]. The above evidence proves that PLK-1 is closely related to the regulation of autophagy.
4.4RoleofPLK-1inepithelial-mesenchymaltransitionThe process by which epithelial cells differentiate into cells with biological characteristics of mesenchymal phenotype according to a certain program is called epithelial-mesenchymal transition (EMT). Numerous pieces of evidence indicate that EMT plays an important role in the progression of many cancers and is a crucial link in invasion and distant spread of tumor cell. There are studies suggesting that PLK-1 may play a role in driving EMT. Overexpression of PLK-1 in prostate epithelial cells can promote carcinogenic transformation and EMT. However, downregulation of PLK-1 activity in metastatic prostate cancer cells can enhance epithelial properties, reverse EMT, and inhibit cell motility. In addition, literature has also shown that PLK-1 can promote the process of EMT in gastric cancer and non-small cell lung cancer[15-16].
5 Correlation between PLK-1 and tumors
PLK-1, as a tumor promoter, plays an important role in mitosis, DNA replication and damage repair, autophagy, and EMT. PLK-1 is overexpressed in various tumors, and its overexpression is often associated with tumor occurrence, invasion, and poor prognosis.
5.1ExpressionofPLK-1ingastriccancerPLK-1 is highly expressed in gastric cancer, and its high expression is associated with the migration and invasion of gastric cancer, as well as a significant reduction in overall survival of gastric cancer patients. Overexpression of PLK-1 can induce centrosome expansion in interphase cells and increase the number of multi-level spindles in mitotic cells, leading to chromosomal missegregation and DNA aneuploidy. In clinical gastric cancer specimens, PLK-1 expression is significantly correlated with the production of DNA aneuploidy, and gastric cancer patients with high PLK-1 expression have poorer prognosis[17].
5.2ExpressionofPLK-1inbreastcancerPLK-1 is also overexpressed in breast cancer, and the overexpression of PLK-1 is related to the low overall survival of breast cancer patients. At the same time, PLK-1 may also be related to the clinical stage of breast cancer. The study found that the expression level of PLK-1 in patients of stage III was higher than that in other stages[18]. Elevated PLK-1 level can also mediate the radioresistance of breast cancer. Some literature shows that it can significantly inhibit the proliferation of breast cancer cells and increase the radiosensitivity of breast cancer cells by inhibiting PLK-1[19].
5.3ExpressionofPLK-1incolorectalcancerThe expression of PLK-1 in colorectal cancer is significantly higher than that in normal colorectal tissue, and its overexpression is associated with recurrence, poor prognosis, and chemotherapy resistance in colorectal cancer. Literature has shown that high expression of PLK-1 is often associated with invasion of colorectal cancer, lymph node metastasis in colorectal cancer patients, and low survival in colorectal cancer patients[20]. The combination of chemotherapy drug oxaliplatin and PLK-1 inhibitor volasertib in colorectal cancer patients can significantly enhance the anti-tumor effect, indicating that it can enhance the chemotherapy effect of oxaliplatin by inhibiting PLK-1[21].
5.4ExpressionofPLK-1inprostatecancerOverexpression of PLK-1 induces EMT like changes in prostate epithelial cells, while knocking down PLK-1 restores the epithelial features of invasive prostate cancer cell lines. In prostate cancer, overexpression of PLK-1 is positively correlated with the grading of prostate cancer. Smad4 is an important tumor suppressor gene in prostate cancer, and PLK-1 can mediate the ubiquitination and degradation of Smad4, thereby promoting the progression of prostate cancer[22].
5.5ExpressionofPLK-1inpancreaticcancerPLK-1 is also associated with the prognosis of pancreatic ductal adenocarcinoma. Studies have found that PLK-1 level is significantly elevated in high-grade pancreatic tumors, and PLK-1 is also associated with pancreatic tumor invasion. Patients with high PLK-1 level have significantly shorter overall survival. The standard chemotherapy drug for pancreatic cancer is gemcitabine, but patients will become resistant to the drug after a period of treatment. Studies have found that the overexpression of PLK-1 mediates gemcitabine resistance in vivo andinvitro[23]. Inhibition of PLK-1invivocan sensitize pancreatic cancer to gemcitabine.
6 PLK-1 inhibitor
PLK-1 controls cell entry into mitosis by mediating the phosphorylation of specific substrates, and is an important regulatory factor in the G2-M phase of the cell cycle. It also participates in DNA damage repair, as well as important biological processes such as autophagy and EMT. Contrary to PLK-l, PLK-2 and PLK-3 have been identified as tumor suppressors to prevent mitotic risk and maintain genomic integrity. The function of PLK-4 is currently not fully understood, but there is evidence to suggest that it may ensure genome stability by regulating centromere replication. PLK-5 is highly expressed in the central nervous system and has been reported as an inhibitor of glioblastoma. Therefore, the optimal strategy for the development of PLK-1 inhibitors is highly selective inhibiting PLK-l activity without inhibiting other PLKs.
PLK-1 inhibitors can exert anti-tumor effects through various mechanisms: blocking the mitotic cycle of tumor cells by inhibiting PLK-1 protein kinase activity, inhibiting tumor cell proliferation, and inducing tumor cell apoptosis; the proliferation of RAS mutated tumor cells depends on key mitotic proteins, and PLK-1 is an important mitotic regulatory factor. Therefore, PLK-1 inhibitors can form synthetic lethality with RAS mutations[24]. PLK-1 is involved in regulating DNA replication and damage repair. Therefore, PLK-1 inhibitors can enhance the anti-tumor effects of other drugs that cause DNA damage, such as chemotherapy drugs. PLK-1 inhibitors can also inhibit tumor angiogenesis by inhibiting the HIF pathway, enhancing the anti-tumor effect of anti angiogenic drugs[25].
According to the structure of PLK-1, there are currently two main types of small molecule inhibitors. One is small molecule inhibitors targeting the PLK-l KD domain, targeting the ATP binding pocket of protein kinases. Most of the PLK-1 inhibitors under study, represented by Onvansertib, belong to this category. Due to frequent mutation in ATP binding pockets, this type of inhibitor typically faces resistance issues. The second is small molecule inhibitors targeting the PLK-l PBD domain, represented by Rigosertib, which have a low binding level with PBD. As of now, at least 10 PLK-1 inhibitors under development have entered clinical trials, but no PLK-1 inhibitors have been launched yet. The summary and introduction of the research progress on some representative PLK-1 inhibitors are as below.
6.1BI2536BI 2536 is a selective small molecule ATP competitive PLK kinase inhibitor developed by Boehringer Ingelheim, and is a representative compound of the first generation of PLK-1 inhibitors. Preclinical studies have shown that BI 2536 can inhibit tumor cell growth in a time/dose dependent manner, induce tumor cell apoptosis, block the tumor cell cycle in the M phase, and inhibit the growth of xenograft tumors in nude mice. Although BI 2536 has achieved a series of successes in preclinical trials, the clinical trial results of BI 2536 are not ideal. In the phase I open dose escalation study (NCT02211859) using BI 2536 alone or in combination with chemotherapy drugs, the tolerability and safety were good. However, phase II clinical trials conducted in patients with relapsed/refractory solid tumors (NCT00526149), non-small cell lung cancer (NCT00376623), and relapsed/refractory acute myeloid leukemia (NCT03303339) showed that BI 2536 lacked anti-tumor activity. This may be related to the short half-life of the drug in the patient’s body and low exposure to tumor cells. Therefore, the clinical trial of BI 2526 as a monotherapy has been terminated, and the development of new PLK-1 inhibitors will continue, with a focus on improving PK properties and increasing the sustained exposure of the drug to tumor tissue.
6.2VolasertibVolasertib, also known as BI 6727, is a small molecule ATP competitive PLK-1 inhibitor obtained by Boehringer Ingelheim by optimizing the structure of dihydropteranone based on BI 2536. Preclinical experiments have shown that volasertib has strong inhibitory activity against various tumors, with anEC50value of 11-37 nmol/L[26]. Compared with BI 2536, volasertib has superior pharmacokinetic properties, such as longer exposure time to tumor drugs, higher tumor distribution and good tissue permeability, longer half-life, and good bioavailability. At present, volasertib alone or in combination with cytarabine (NCT00804856) and docetabine (NCT02003573) has entered phase II clinical trials for acute myeloid leukemia, and encouraging results have been achieved in tumor treatment. However, clinical trials conducted in patients with advanced or metastatic non-small cell lung cancer (NCT00824408), refractory ovarian cancer (NCT01121406), and advanced or metastatic urothelial carcinoma (NCT01023958) have shown that volasertib only shows moderate efficacy.
6.3OnvansertibOnvansertib, also known as NMS-P937, is a novel PLK-1 inhibitor developed by Cardiff Oncology. Unlike previous PLK-1 inhibitors, its parent nucleus structure is pyrazoquinazoline, which can be administered orally without the need for intravenous administration. Preclinical studies have shown that Onvansertib has broad-spectrum anti-tumor proliferative activity, and bothinvitroandinvivoanti-tumor activity is demonstrated when used alone or in combination with other anti-tumor drugs. The standard treatment (SOC) for RAS mutated metastatic colorectal cancer is a combination of chemotherapy drug FOLFOX/FOLFORI and anti angiogenic drug bevacizumab. In 2019, Cardiff Oncology firstly launched the second-line treatment of RAS mutated metastatic colorectal cancer using the combination of Onvansertib and SOC (NCT03829410, NCT05593328, NCT04446793). Preliminary research results have found that in patients who have never received bevacizumab treatment, the objective response rate (ORR) after Onvansertib and SOC treatment is as high as 73%, while the historical ORR is only 23%-26%. Patients who received bevacizumab treatment had an ORR of only 16% after Onvansertib and SOC treatment. Therefore, Cardiff Oncology has initiated a phase II clinical trial (NCT06106308) of Onvansertib combined with SOC for first-line treatment of RAS mutated metastatic colorectal cancer, with an expected start date of November 16, 2023 and completion date of January 2027. The experiment was divided into 6 groups: Onvansertib 20 mg+SOR (FOLFIRI+bevacizumab) group, Onvansertib 30 mg+SOC (FOLFIRI+bevacizumab) group, SOC (FOLFIRI+bevacizumab) group, Onvansertib 20 mg+SOR (FOLFOX+bevacizumab) group, Onvansertib 30 mg+SOC (FOLFOX+bevacizumab) group, and SOC (FOLFOX+bevacizumab) group, to examine the therapeutic effects of different doses of Onvansertib, different chemotherapy regimens, and the combination of bevacizumab.
In addition, other clinical trials being carried out by Onvansertib are: phase II clinical trial of Onvansertib combined with Nal-IRI/5-fluorouracil/folic acid for second-line treatment of metastatic pancreatic ductal adenocarcinoma (NCT04752696), phase II clinical trial of Onvansertib combined with paclitaxel for second-line treatment of triple negative breast cancer (NCT05383196), phase II clinical trial of Onvansertib in combination with low-dose cytarabine or dexamethasone for the treatment of adult acute myeloid leukemia (NCT03303339), phase II clinical trial of Onvansertib in combination with abiraterone and prednisone for the treatment of metastatic castration resistant prostate cancer (NCT03414034).
6.4GSK461364GSK461364 developed by GlaxoSmithKline and Onvansertib are both the third generation of PLK-1 inhibitors. The structure of GSK461364 is imidazolium triazine, which is an ATP competitive and highly selective PLK-1 inhibitor. In preclinical experiments, GSK461364 can widely inhibit cancer cell proliferation, induce cancer cell apoptosis, and inhibit the growth of xenograft tumors in nude mice, whether used alone or in combination with other anti-tumor drugs. GSK461364 has good preclinical safety and anti-tumor activity, and has now progressed to clinical phase I (NCT00536835). In phase I clinical trials, the adverse reactions of GSK461364 are mild, mainly including neutropenia, fatigue, nausea, venous thromboembolism,etc.There have been no reports of GSK461364 entering phase II clinical trials.
6.5TAK-960TAK-960 is a pyrimidine diazepinone analogue developed by Takeda in 2012, which inhibits PLK-1 by targeting the ATP binding domain. It is an orally effective selective PLK-1 inhibitor. In preclinical trials, TAK-960 has been proven to be an effective drug for the treatment of colorectal cancer. In the phase I clinical trial targeting solid tumors (NCT04831502), TAK-960 showed poor therapeutic efficacy and severe adverse reactions. Therefore, Takeda terminated the clinical trial of TAK-960.
7 Conclusions and prospects
PLK-1 is the main regulatory factor of mitosis, and plays various important roles in mitosis, including controlling the progression of mitosis, centrosome maturation and separation, spindle assembly, and cytoplasmic division and shedding. Meanwhile, PLK-1 also plays an important role in DNA replication, damage repair, and autophagy. The high expression of PLK-1 in tumor cells may lead to chromosomal instability, dysregulation of multiple carcinogenic pathways, and promote EMT. Therefore, overexpression of PLK-1 is often associated with poor prognosis and tumor invasiveness, which drives the development of inhibitors targeting PLK-1.
Although PLK-1 inhibitors have shown good anti-tumor effects in preclinical trials, only a few drugs have shown therapeutic effects in clinical trials, such as Onvansertib combined with FOLFIRI/bevacizumab showing good therapeutic effects in RAS mutant colon cancer patients who did not receive bevacizumab treatment. The significant difference between Onvansertib and other PLK-1 inhibitors lies in its higher selectivity towards the target PLK-1. High target specificity can reduce off target effects, reduce adverse drug reactions, and use higher drug doses in clinical practice, thereby improving clinical treatment outcomes.
PLK-1 inhibitors can exert anti-tumor effects through various mechanisms, including blocking the mitotic cycle of tumor cells, forming synthetic lethality with RAS mutations, enhancing the anti-tumor effects of chemotherapy drugs and anti angiogenic drugs. Therefore, the use of PLK-1 inhibitors alone or in combination with chemotherapy/anti angiogenic drugs is expected to become an important treatment method for KRAS mutant tumors.
杂志排行

Medicinal Plant的其它文章
- Effects of Exogenous Plant Hormones on Growth Status and Secondary Metabolism of Houttuynia cordata Thunb.
- Identification of Xunxi Shujin Decoction by TLC
- Preparation of 20 (S)-protopanaxadiol PLGA Nanoparticles
- A Network Pharmacology Study on Active Components and Targets of Citri Reticulatae Pericarpium for Treating Keloids
- Pharmacognostic Identification of Hedyotis auricularia and Mitracarpus villosus
- Determination of Salvianolic Acid B in Yiqi Huayu Prescription by HPLC