AMME chromosomal region gene 1基因变异矮小相关综合征一例及文献复习
2024-03-07王小红杨海花高静陈永兴卫海燕
王小红,杨海花,高静,陈永兴,卫海燕
[ 郑州大学附属儿童医院(河南省儿童医院郑州儿童医院)内分泌遗传代谢科,河南 郑州 450000 ]
Alport 综合征、智力缺陷、面中部发育不全和椭圆形红细胞增生症(Alport syndrome,mental retardation,midface hypoplasia,and elliptocytosis,AMME)是一种与Xq22.3q23 区域微缺失相关的连续基因综合征[1-5],AMME 染色体区域基因1(AMME chromosomal region gene 1,AMMECR1)是AMME 关键区域基因之一[6]。国外学者报道AMMECR1基因变异可引起发育延迟/智力缺陷、中面发育不全、耳聋、矮身材和椭圆形红细胞增多症等AMME 综合征大多数临床特征,不发生Alport 综合征[7-9]。目前,国内鲜见该病报道。本文报道了1 例AMMECR1基因新变异患儿的临床表现及基因特点,并通过复习相关文献归纳总结其特征。
1 病例介绍
1.1 研究对象
患儿,男,2 岁8 个月,主因“生长发育迟缓2 年”于2021 年5 月25 日入院。8 个月后出现生长迟缓,出生身长49 cm(-0.8 SD),出生体重3.3 kg(0.0 SD),1 月龄身长56 cm(0.7 SD),体重4.1 kg(-0.2 SD),2 月龄身长56.5 cm(-1.0 SD),体重4.5 kg(-1.9 SD),3 月龄身长62 cm(0.1 SD),体重6.3 kg(-0.6 SD),8.5 月龄,身长66 cm(-2.0 SD),体重8 kg(-0.2 SD),13.5 月龄身长70.5 cm(-2.2 SD),体重9 kg(-1.3 SD)。平素挑食,食欲差,余无不适。患儿系G1P1,孕40 周顺产。生长发育史:2 个月会抬头,1 岁会独坐,1 岁6 个月会爬、会独站,1 岁7 个月会走。语言发育:目前会发叠音字。既往史、家族史无异常。父亲身高:165 cm,母亲身高:158 cm。入院查体:身长79.3 cm(-4.0 SD),体重9.5 kg(-3.0 SD),头围45 cm,体重指数(BMI)15.11 kg/m2,神志清,精神好,全身皮肤无黄染、皮疹及出血点,前额扁平,面部扁平,鼻梁低平,耳大,上唇薄,小下颌,心肺腹及神经系统查体无明显异常。阴茎长4 cm,双侧睾丸1 mL,阴毛Tanner 1 期。患儿生长发育曲线见图1。

图1 患儿生长曲线图
1.2 实验室检查
血常规:血红蛋白110 g/L,红细胞3.91×1012/L,血细胞比容35.1%,余正常。尿常规:比重1.010,酸碱度7.0。胰岛素样生长因子41.201 ng/mL(50~300 ng/mL),基础生长激素6.6 ng/mL,25 羟维生素D 42.5 ng/mL,甲状旁腺激素51 pg/mL,骨源性碱性磷酸酶250 U/L。肝肾功能、电解质、糖化血红蛋白、甲状腺功能、肾上腺功能、血氨基酸及酰基肉碱谱、尿有机酸分析:正常。心电图、心脏彩超、肝胆脾肾彩超、垂体MRI 无异常,听力筛查:正常。考虑患儿年龄偏小,家属拒绝X 线检查。在其父母知情同意并签署书面同意书,并经医院伦理审查委员会审查批准后(伦理号:2021-K-047),收集患儿及其父母外周血标本各2 mL,EDTA 抗凝,提取基因组DNA,构建基因组文库,进行全外显子基因测序,检出X 染色体上AMMECR1基因第2 外显子c.474C>A:p(Y158X)半合变异,母亲携带无义变异c.474C>A:p(Y158X),父亲为野生型(图2)。c.474C>A 为新变异,经检索千人基因组、ESP6500 及dpSNP 数据库基因变异数据库和相关文献为未报道过的新变异。c.474C>A 变异导致AMMECR1基因的第158 位密码子由编码酪氨酸变为终止密码子,该变异为无义变异,可能导致蛋白质功能缺失。该变异在正常人群数据库频率为未收录,HGMD 数据库无该位点的相关性报道,Clinvar 数据库无该位点的致病性分析。根据ACMG 指南,该变异初步判定为可能致病性变异(PVS1+PM2)。
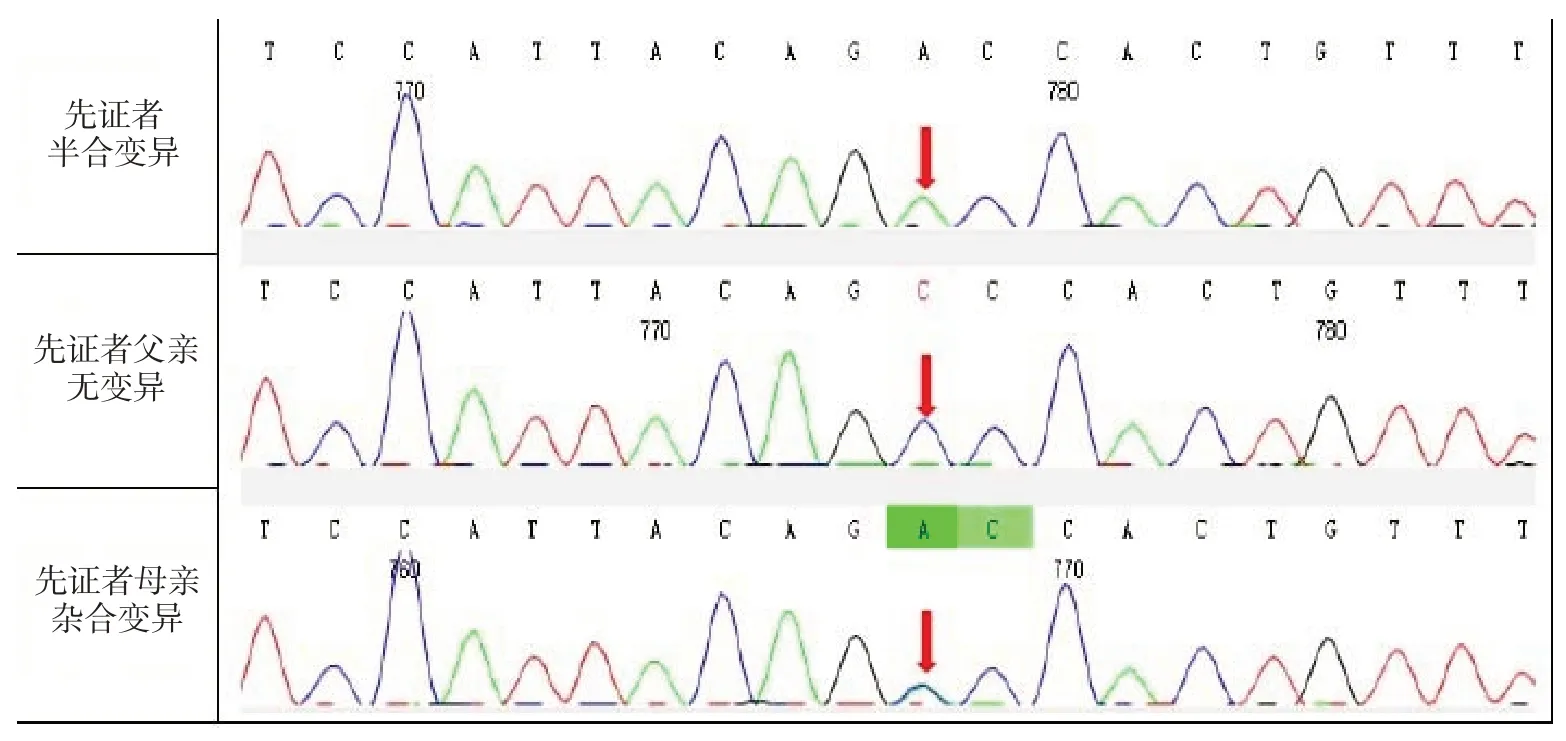
图2 AMMECR1 基因测序结果
1.3 治疗及随访
入院后予饮食指导,建议合理喂养,保证每日蛋白质及总热量供应,保证充足睡眠,加强纵向运动,给予补充维生素D 滴剂800 U/d,补充碳酸钙D3颗粒3 g/d。同时建议应用生长激素,但患儿家属拒绝。患儿3 岁7 月龄复诊,智力及运动发育正常,会说词语,身高82.9 cm(-3.6 SD),体重11 kg(-2.8 SD)。血常规:血红蛋白120 g/L,红细胞4.07×1012/L,血细胞比容35.4%,余正常。尿常规:比重1.010,酸碱度6.5。胰岛素样生长因子42.55 ng/mL,25 羟维生素D 48.12 ng/mL,甲状旁腺激素46 pg/mL。尿钙肌酐比0.75(<0.2),肾小管功能:尿液α1微球蛋白12.3 mg/L(<5 mg/L)、尿液β 微球蛋白2 mg/L(<0.3 mg/L)、尿-N-乙酰-β-D-氨基葡萄糖苷酶10.2 U/L(<12 U/L)。尿微量白蛋白8.1 mg/L。肝肾功能、血气分析、电解质、糖化血红蛋白、甲状腺功能、肾上腺功能:正常。心脏彩超、肝胆脾肾彩超及听力筛查正常。患儿骨龄、下肢骨骼片未见明显异常(图3)。

图3 患儿双下肢骨骼及骨龄片
2 讨论
染色体Xq22.3-Xq23 区域包括整个COL4A5基因及其延伸到端粒的邻近基因:GUCY2F、NXT2、KCNE1L、ACSL4、TMEM164、MIRM978、AMMECR1、SNORD96B、RGAG1、TDGF3、CHRDL1、PAK3和DCX[8]。该区域的缺失可能出现Alport 综合征,并伴有智力障碍、中面部发育不全和椭圆形红细胞增多症、发育迟缓、语言迟缓以及骨骼、心脏和眼睛的改变,表型的差异与基因缺失的位置和大小有关。
AMMECR1基因在AMME 复杂区间内,它定位于染色体Xq23[6],全长246.13 kb,包含6 个外显子。AMMECR1基因的C 端区域高度保守,同源物出现在从细菌、古细菌到真核生物的物种中。AMMECR1结构域的高度保守表明其具有基本的细胞功能,可能是在转录、复制、修复或翻译机制中发挥作用[8-12]。编码蛋白含4 种亚型,长度分别为333、296、210、319 个氨基酸[13]。编码蛋白具有假定的核定位信号功能,被命名为AMMECR 核蛋白1[14]。目前AMMECR1基因编码蛋白功能尚不明确。ANDREOLETTI 等[8]研究显示,AMMECR1基因单点突变引起特异性蛋白功能障碍,导致面中部发育不全和椭圆形红细胞增多,导致早期语言发育迟缓、婴儿张力减退和听力损失,可能在畸形、肾钙质沉着和黏膜下腭裂中发挥作用。MOYSÉS-OLIVEIRA 等[9]研究AMMECR1基因敲除的斑马鱼与患者出现相似的临床表现,且表型的严重程度随基因敲除的程度而增加,提出AMMECR1基因失活与涉及生长、骨骼表型及心脏改变的新综合征相关。其基因突变患者一般在儿童早期发病,表型与AMME 综合征有重叠,但是无Alport 综合征表现。
分别以“AMMECR1gene”“Xq22.3-q23 deletion”为检索词,检索建库至2023 年6 月30 日为止万方、维普、中国知网CNKI 数据库相关文献,均未发现相关报道;再次以“AMMECR1gene”“Xq22.3-q23 deletion” 为检索词检索PubMed 数据库,从7 篇英文文献中获得涉及AMMECR1基因变异的临床病例20 例,来自13 个家系,男15 例(其中1 例为胎儿),女4 例,未知性别胎儿1 例。其中变异类型包含染色体平衡易位、基因缺失、点突变。因临床表型与基因缺失片段大小有关,故本文主要复习AMMECR1基因单位点突变。文献复习及本研究共报道7 例AMMECR1基因单位点突变,包含错义突变和无义突变,既往报道2 例c.G530A(p.G177D)错义突变,1 例c.502C>T(p.Arg168Ter)无义突变,1 例c.429T>A(p.Tyr143Ter)无义突变,2 例c.C133T(p.R45X)无义突变。所有患儿均为男孩,诊断年龄从11 个月至8 岁不等,其中7 例患儿存在身材矮小及发育迟缓,6 例存在早期婴儿肌张力减低及听力损失,5 例存在运动语言落后及面中部发育不全,4 例存在心脏改变(房缺、卵圆孔未闭、动脉导管未闭),3 例存在腭裂、骨骼改变及椭圆形红细胞增多症,2 例存在智力落后和肾钙质沉着症。本研究发现患儿存在X 染色体上AMMECR1基因第二外显子c.474C>A(p.Y158X),无义突变。因过早引入终止密码子,导致无意义介导的mRNA衰变和/或蛋白质截断,从而影响蛋白功能。本例患儿表现出面中部发育不全,婴儿期肌张力减低,身材矮小,运动及语言发育迟缓,随访中出现肾小管功能受损,尿钙肌酐比增高。身材矮小是每个患儿都有的,而其他临床表现并不是每个患儿均具有,说明本病的临床异质性大。而本例患儿在随访中出现新的症状,提示本病需要长期随访观察。
目前该病的治疗主要为对症治疗。身高方面,未见AMMECR1基因单碱基突变患儿应用生长激素治疗的相关报道。文献复习共观察到3 例Xq22.3-q23 微缺失患儿运用生长激素治疗[7,9],其中有2 例患儿对生长激素治疗有反应,1 例为女孩:46,X,t(X;9)(q23;q11.2)平衡易位,其对生长激素治疗有反应,但具体随访无相关资料,1 例为男孩,身高从4 岁时<2.5 SD 到11 岁时130 cm(10th);另1 例男孩无反应,但缺乏相关随访资料。本例患儿建议其予生长激素治疗,但家属拒绝。其余表型的改变应尽早实施干预,以便改善患儿后期生活质量,早期诊断可改善患儿预后。
综上所述,AMMECR1基因c.474C>A(p.Y158X)半合变异是患儿的主要致病原因,基因测序结果为临床诊断及遗传咨询提供了依据。
