磷化电流密度对A286 合金电化学磷化膜组织结构和结合强度的影响
2024-03-06杜孟飞曾宇乔张旭海
胡 勇, 周 昊, 杜孟飞, 曾宇乔, 张旭海, 方 峰
(1.东南大学材料与科学工程学院, 江苏 南京 211189; 2.宝钢金属有限公司, 上海 200940)
0 前 言
A286 合金是一种Fe-25Ni-15Cr 基合金,在高温条件下能表现出高的屈服强度和持久的蠕变强度以及良好的焊接性能,常用于制造650 ℃以下长期工作的高温承力部件,如发动机转子、耐热螺栓和涡轮叶片等[1]。 但A286 合金在冷拉拔、冷墩等过程中,易出现表面损伤,给后期零部件的使用造成安全隐患。
化学磷化工艺可以很好地解决金属在冷加工过程中出现的表面损伤问题[2]。 其工艺过程主要是通过化学反应在金属表面形成磷酸盐转化膜(磷化膜)[3]。 磷化膜具有显著的减摩和润滑作用[4],可在冷加工过程中为基体金属提供良好的保护,目前已在许多金属材料的表面防护中获得应用[5]。 但A286 合金中含有大量Ni、Cr 元素,在开路电位下,合金表面易在镀液中形成稳定的钝化膜,难以通过普通化学磷化工艺在其表面沉积出高膜基结合力的薄膜[6]。 针对这一问题,研究人员通过氮化[7]、水热法[8]、热浸镀锌[9]、电耦合[10]等方法来提高A286 合金表面的电化学活性,从而可以顺利完成后续的磷化处理,但这些方法存在工艺复杂、耗时长和温度高等诸多问题,很难在实际大规模生产中应用。
电化学磷化(电磷化)是在合金上施加一个较大的阴极电流/电压,一方面可以防止合金腐蚀或钝化,另一方面可以使合金表面发生析氢反应[11]。 当合金附近区域的H+浓度降低时,磷酸盐的过饱和度提高,便可在合金上沉积出磷化膜[12]。 采用该工艺在不锈钢、钛和铝等耐蚀金属表面均可进行磷化[13-18],但尚未见到在A286 高温合金上成功应用的报道。
本工作对A286 合金进行表面电化学磷化处理,通过调节磷化电流密度,优选出了成膜致密、膜基结合力高的A286 高温合金磷化工艺,明确了磷化膜在不同电流密度条件下的成膜机制,为电化学磷化工艺在高温合金上的实际应用提供了理论依据。
1 实验材料与方法
磷化基体为A286 合金。 磷化前,先将A286 合金片依次用600,1 000,1 200 号砂纸打磨,再进行超声碱洗(20 g/L NaOH +3 g/L OP-10 乳化剂)和酸洗[10%(体积分数)HNO3+3%(体积分数)HCl]。 各工序之间,样品用去离子水充分清洗。 电化学磷化采用三电极体系,以A286 合金(1 cm2)为工作电极,铂片(225 cm2)为对电极, 饱和甘汞电极(SCE)为参比电极。 沉积模式为恒流(阴极电流密度20~160 mA/cm2,电流密度<20 mA/cm2时,无明显析氢反应;电流密度>160 mA/cm2时, 对电极易氧化溶解),沉积时间600 s,沉积温度75 ℃。 磷化液由南京派诺公司提供,其主要参数为:总酸度(TA)为155.2,游离酸度(FA)为22.8,主要成膜离子(Zn2+)浓度为50 g/L。 磷化后的样品分别用去离子水、无水乙醇清洗并吹干。
采用Sirion 场发射扫描电镜(SEM)观察磷化膜的表面形貌。 用能谱仪(EDS)分析试样的化学成分。 用X 射线衍射仪(XRD)分析磷化膜的相组成情况。 用自动划痕仪(WS-2005)检测膜基结合力,划痕速度为1 mm/min,加载速度为10 N/min,最佳加载为60 N。
2 结果与讨论
电化学磷化过程中工作电极上电位随时间的变化曲线(φ-t曲线)如图1 所示。 无论电流密度大小如何,样品表面的φ均随t呈先急剧负移、再在某个电位附近小幅振荡的变化规律。φ负移与电极表面的浓差极化有关。 当电极上施加的电流密度达到析氢条件时,2H++2e→H2反应进行,样品表面的H+浓度迅速降低,会导致析氢过电位增加、φ负移。φ在某个电位小幅震荡,则是析氢、磷化及离子扩散达到动态平衡的结果。随着磷化电流密度的增加,准稳态的φ值负移,且震荡加剧,这是析氢反应加剧的表现。
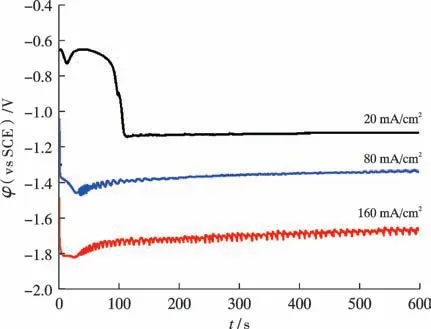
图1 不同磷化电流密度条件下,磷化过程的φ-t 曲线Fig.1 φ-t curve of phosphating process under different phosphating current densities
为探究磷化电流密度对磷化膜沉积机制的影响,本研究选取了各阴极电流密度下沉积不同时间的样品,对其表面形貌和成分进行比较。 磷化电流密度为20 mA/cm2时,样品表面形貌随时间的变化如图2 所示。 磷化15 s 后,样品表面尚未形成沉积物(图2a)。磷化30 ~60 s 时,样品表面开始出现少量沉积物(图2b),EDS 及XRD 分析表明其主要成分为Zn3(PO4)2·4H2O。 这些Zn3(PO4)2·4H2O 晶体的尺寸约为50 ~100 μm,大部分呈叶片状(图2b 插图及2c 插图),只有少量呈针状。 这表明晶体生长为扩散-激活混合控制型,结晶驱动力不高[19]。 120 s 后,磷化开始进入φ-t曲线对应的第二阶段。 此时,针状Zn3(PO4)2·4H2O晶体的数量有显著增加(图2d)。 这是由于析氢反应消耗了样品附近大量的H+,Zn3(PO4)2·4H2O 结晶驱动力增加。 磷化至300 s 时,样品表面已基本被Zn3(PO4)2·4H2O 覆盖。 此时,可以观察到大量的针状及片状Zn3(PO4)2·4H2O 晶体(图2e)。 磷化600 s后,样品最表层为等轴片状Zn3(PO4)2·4H2O,尺寸约为50 μm(图2f)。

图2 磷化电流密度为20 mA/cm2时,样品表面形貌随磷化时间的演变过程Fig.2 Evolution of sample surface morphology with the phosphating current density of 20 mA/cm2 when the phosphating time
磷化电流密度为80 mA/cm2时,样品表面形貌的演变过程如图3 所示。 磷化5 s 后,合金表面出现少量针状Zn3(PO4)2·4H2O(图3a)。 磷化15 s,针状沉积物数量显著增加(图3b)。 针状晶的大量析出,表明在80 mA/cm2条件下,Zn3(PO4)2·4H2O 的晶体生长转变为扩散控制性, 结晶驱动力较高。 这些针状Zn3(PO4)2·4H2O 在A286 合金表面多从一点出发,向面内四周放射生长,呈直径为30~50 μm 的花状。 30 s后,磷化进入φ-t曲线对应的第二阶段。 花状Zn3(PO4)2·4H2O 长大,直径增至150 ~200 μm (图3c)。 值得注意的是,在Zn3(PO4)2·4H2O 之间,出现了直径为5 μm 的圆环状沉积物(图3c 插图)。 EDS 成分分析表明其主要成分是Zn。 此后,随磷化反应的持续推进,金属锌被大量直径为3 ~5 μm 的绒球状沉积物覆盖(图3d)。 EDS 分析表明,其主要成分也是Zn3(PO4)2·4H2O。 300 s 后,样品最表面被棒状Zn3(PO4)2·4H2O 完全覆盖,未再观测到金属Zn。 磷化600 s 后,最表层Zn3(PO4)2·4H2O 形态无明显变化(图3f)。

图3 磷化电流密度80 mA/cm2,样品表面形貌随磷化时间的演变过程Fig.3 Evolution of sample surface morphology with the phosphating current density of 80 mA/cm2 when the phosphating time
图4 为磷化电流密度160 mA/cm2、不同沉积时间的样品表面形貌。 磷化5 s 时,样品表面已被大量细小的球状Zn 覆盖(图4a),其直径约为1 μm (图4a 插图)。 15 s 时,在Zn 间形成网状分布的Zn3(PO4)2·4H2O (图4b)。 从30 s 到60 s,随着磷化过程的推进,Zn 逐渐被Zn3(PO4)2·4H2O 覆盖(图4c,4d 及插图),表明Zn3(PO4)2·4H2O 一直呈绒球状,直径2 ~3 μm。但磷化至300 s 时,样品最表面的沉积层为均匀的短棒状Zn3(PO4)2(图4e);且随着磷化时间的延长,棒状磷酸锌晶体逐渐细化,表面变得更加致密、均匀(图4f)。

图4 磷化电流密度160 mA/cm2,样品表面形貌随磷化时间的演变过程Fig.4 Evolution of sample surface morphology with the phosphating current density of 160 mA/cm2 when the phosphating time
XRD 检测结果显示,沉积600 s 后所获的磷化膜均以Hopeite Zn3(PO4)2·4H2O(JCPDS#37-0465)为主相(图5)。 随着磷化电流密度的增加,Hopeite 的(020)和(220)衍射峰强度显著降低,表明阴极电流密度对Zn3(PO4)2·4H2O 的择优取向具有显著影响。 当阴极电流密度高于20 mA/cm2时,沉积样品中的Hopeite 相(220)衍射信号远低于Hopeite 的粉末衍射结果(JCPDS#37-0465),表明磷化膜中的针状、棒状Hopeite 在样品面内的择优取向可能为(220)。
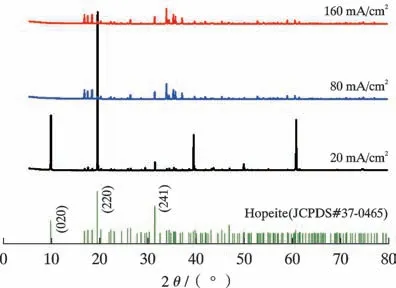
图5 不同磷化电流密度条件下所获磷化膜的XRD 谱Fig.5 XRD images of phosphating film obtained under different phosphating current densities
综合SEM、EDS 和XRD 实验结果可知,A286 合金表面的电化学磷化过程具有以下几个特点:
(1)沉积过程中,低阴极电流密度条件下,沉积物为单一的Hopeite;阴极电流密度较高时,沉积顺序为Hopeite→Hopeite+Zn→Hopeite;高阴极电流密度条件下,沉积顺序则为Zn→Hopeite+Zn→Hopeite。 初期沉积产物由Hopeite 向Zn 转变,可能是由于样品表面电位随电流密度增加而负移,达到了Zn 的析出电位。 而后续的沉积相转变为Hopeite,是由于Zn3(PO4)2·4H2O的大量沉积会降低表面Zn2+浓度,导致Zn 的析出电位负移,Zn2++2e→Zn 反应变得更困难。
(2)磷化膜在A286 合金上的沉积不是Hopeite 晶体由基体向面外择优生长的过程,而是以沉积物逐层生长、铺叠的方式进行。 此外,Hopeite 在平行于基体的方向上存在择优取向。 这种生长模式与Hopeite 相析出的机制有关。 Hopeite 析出主要受过饱和度影响[20]。在阴极电流作用下,A286 合金表面发生析氢反应,在垂直于合金表面的方向上存在较大的H+浓度梯度。而合金表面的H+浓度最低,Zn2+和PO43-过饱和度最高,Hopeite 相更倾向于在面内择优生长。
(3)增加磷化电流密度,可促进沉积产物的组织细化[21]。 这是由于高的阴极电流可提升析氢的反应速率,使样品表面Zn2+和PO43-的过饱和度增大,Hopeite相形核率增加,晶体尺寸细化[22-24]。
图6 是磷化后样品进行划痕试验后的表面SEM 形貌。 在20 mA/cm2条件下沉积的磷化膜,与A286 合金基体间的膜基结合力为6 N。 由图可知,压头加载至6~9 N 时,划痕周围已出现明显的开裂区(图6a 方框)。 在EDS 面扫分析中,开裂区富Fe (图6d),贫Zn(图6f)、贫P(图6h),表明磷化膜已完全剥离。 随磷化电流密度的增加,磷化膜剥离的临界载荷提高。 磷化电流密度为80 mA/cm2时,压头加载至35 N 出现完全剥离区(图6b 方框)。 磷化电流密度为160 mA/cm2时,压头加载至60 N 也未观察到明显的开裂情况。EDS 面扫结果显示,压头划过该磷化膜的区域(图6c方框)贫P(图6i)、但Fe(图6e)和Zn(图6g)均匀分布,表明加载至60 N 时,尽管Hopeite 相被剥离,但Zn相仍与A286 合金保持良好的结合。

图6 不同磷化电流密度条件下所获磷化膜划痕试验后样品表面SEM 形貌Fig.6 SEM images of sample surface after scratch test of phosphating film obtained under different phosphating current density conditions
图7 为不同电流密度下制备的磷化膜结合力对比图。 由图7 可知,在160 mA/cm2条件下制备的磷化膜膜基结合力最高,具有较好的耐磨性能。 这与膜基处存在少量Zn 沉积物及Hopeite 相的组织结构细小、致密有关。 Zn 较软,可以提供良好的润滑性,而Hopeite晶体细小、结构致密,可以提高磷化膜的强度[25]。
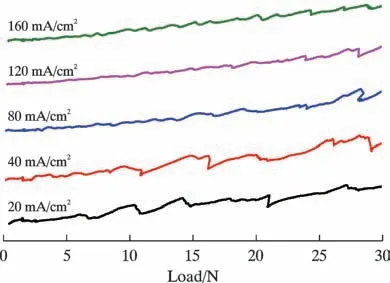
图7 不同磷化电流密度条件下所获磷化膜结合力对比图Fig.7 Comparison of binding force of phosphating film obtained under different phosphating current density conditions
3 结 论
利用电化学磷化可以在A286 合金表面沉积出磷化膜。 磷化电流密度对磷化膜的相组成、微观组织结构和摩擦学性能均有显著影响。 低电流密度下,A286合金表面沉积出的磷化膜为单一的Hopeite 相,组织结构粗大、疏松,膜基结合力为6 N。 提高磷化电流密度,可使磷化膜相结构转变为Zn+Hopeite 复相。 膜层组织结构细化、致密,膜基结合力显著提高。 当电流密度达到160 mA/cm2,膜基结合力高于60 N。
