基于多重长PCR靶向捕获测序技术的高同源SNP鉴定
2024-02-29王决恒周宇荀肖君华
王决恒,周宇荀,李 凯,肖君华
(东华大学 化学化工与生物工程学院, 上海)
单核苷酸多态性(single nucleotide polymorphism,SNP)指基因组DNA序列中的单碱基(A,T,C和G)突变引起的DNA片段多态性[1-2],其位点的变化类型包括碱基的插入、缺失、倒置等。通常采用Sanger测序、SNP shot技术、PCR-LDR分型技术、TaqMan探针法、高通量测序法等[3-4]检测SNP,其中多重PCR(polymerase chain reaction)捕获二代测序[5]因具有高效率、高通量、低成本的特性而被广泛应用于大规模SNP分型领域[6-8]。但对高同源区段的SNP进行分型时,由于同源片段的影响,靶向高通量测序难以准确地进行SNP分型[4,9]。
高同源区段SNP是指SNP所在200~400 bp片段(SNP位于片段中间位置)在其基因组上同源性超过80%、同源序列数量≥2。作为比较特殊的SNP类型[8-9],高同源SNP的准确分型将是一个巨大的挑战。为避免试验前期浪费时间和原材料,应在试验前先采用生信技术预评估SNP所在片段的同源性,并将同源性较高的片段筛选出来进行测序。因此,需要通过BLAST(basic local alignment search tool)数据库将高同源片段与基因组数据库进行全序列比对[10],标记出延长引物覆盖范围(1~4 kb)后的SNP两端的特异区域,然后在特异性区段上设计特异性多重长PCR引物,并借助多重长PCR技术靶向捕获目标片段,避免其他同源片段的干扰。最后,进行第二次靶向扩增子捕获并建库测序。二次多重扩增子捕获应通过定量PCR技术调节多重扩增子表达量一致,从而保证多重扩增子文库的均一性。
本研究利用多重长PCR反应[11-12]对329个样本进行9个高同源区段SNP分型,先通过6重长PCR靶向捕获9个高同源SNP所在片段,再通过8重(有2个SNP处于同一扩增子片段)扩增子捕获建库,最后应用HWE法则计算二代测序数SNP的基因频率。为了进一步确认分析结果的准确性,使用本实验室PCR-LDR(ligase detection reaction)技术探索得到的经验性阈值进行验证,记等位基因比率在15%~85%的位点为杂合子,在此范围之外的为纯合子[13]。若两次计算结果一致,再将其分型结果与NCBI数据库基因频率进行比较并计算HWE对应p值[14-17],判定测序结果是否可靠。本研究有望提出一个准确、高效的高同源SNP分型方案。
1 材料与方法
1.1 DNA提取
本试验的329个血样样本均来自江苏省太湖干部疗养院志愿者(所有参与志愿者均已知情);采用磁珠法通用型基因组DNA提取试剂盒(天根生化科技(北京)有限公司,货号:DP705);DNA提取操作步骤依说明书进行,并以0.8%琼脂糖凝胶电泳和NanoDrop2000C型超微量分光光度计(Thermo Fisher Scientific,美国)对DNA进行质控。
1.2 SNP同源性评估
在Bio-Linux 8.04平台构建本地BLAST数据库[18](使用人类基因组版本为hg19)后,打开Terminal输入代码blastn-query input_nuclear.fasta-db database_hg19-out output.xlsx-outfmt 6得到评估结果。Input_nuclear.fasta表示需要同源性评估的序列,database_hg19作为比对的参考基因组数据库,output.xlsx表示输出结果。将评估结果中高同源区段的SNP片段筛选出来,通过设计长片段特异性引物捕获该区段。
1.3 引物设计
9个SNP位点的rs号和基因序列信息来源于美国NCBI(national center for biotechnology information)。所有特异引物用Oligo7.6软件设计,特异引物设计遵循规则包括解链温度(tm)为55~65 ℃,GC含量为40%~60%,与Blast比对后,多重长PCR的上、下引物位置应设置在序列唯一性较好的位置,如图1所示。由图1可知:在特异区A设计上游引物,特异区B设计下游引物;特异区的选择标准为尽可能避开红色、紫色、绿色、蓝色区域,Alignment Scores数值越小越好。

图1 NCBI比对Graphic Summary结果中标记的特异性较好的位置Fig.1 NCBI compared with Graphic Summary results to mark the position with better specificity
多重长PCR靶向捕获扩增子建库二代测序方法:一轮多重长PCR特异性引物无需通用接头(见表1);二轮多重PCR特异引物上游和下游分别添加通用接头序列,上游通用序列5′-ACGACGTGTCG-AGTTCAGGT-3′,下游通用序列5′-CAGTGAGT-CGCCACAGGTCA-3′(见表2)。为标记不同样本,以携带不同index序列的接头引物标记不同样本,329个样本分别由不同的接头引物标记。所有设计的多重引物均使用Oligo 7.6和Primer 3-py软件进行引物互作分析,将不会发生互作的引物设为同一组构成多重PCR。所有引物皆由上海赛索飞生物科技有限公司合成。所有引物和探针皆由上海赛索飞生物科技有限公司合成。

表1 多重长PCR引物Table 1 Multiplexed long PCR primers
由表1可知,LP为多重长PCR引物名称,扩增长度为1.4~3.4 kb。使用Oligo 7.6及Primer 3-py软件对6对长PCR引物进行引物互作分析,结果显示6对引物不会发生互作现象,可以构建6重长PCR反应。由表2可知,rs ID为SNP对应的rs号,ACGACGTGTCGAGTTCAGGT作为上游引物的通用序列、CAGTGAGTCGCCACAGGTCA作为下游引物的通用序列。添加通用序列是为了便于第三轮PCR扩增子建库添加Index、I5/I7。表中rs代表引物名称,扩增长度为183~220 bp。8对扩增子引物(rs28399468、rs5031016共用一对扩增子引物)使用Oligo 7.6及Primer 3-py软件进行引物互作分析,结果显示8对引物不会发生互作现象,因此可以构建8重扩增子反应。
1.4 扩增子测序文库构建
目标序列的靶向精准捕获是检测高同源SNP位点的基因频率的关键,保证多重PCR产物均一性是准确鉴定SNP的基因频率的重要标准,通过荧光定量PCR法(染料法)调试第二轮PCR引物浓度,保证每对引物均匀扩增。每轮PCR产物均进行磁珠纯化、稀释,尽可能消除上一轮PCR反应体系中模板、引物的残留,避免下一轮PCR受影响。3轮多重长PCR靶向捕获扩增子建库测序技术原理示意图如图2所示。由图2可知:特异引物包括特异序列(白色)和通用序列(黑色)两个部分;接头引物包括接头序列、特异index序列和通用序列等3部分。

图2 多重长PCR靶向捕获扩增子建库测序技术原理Fig.2 Principle of multi-long PCR targeted capture amplicon library construction and sequencing technology
多重长PCR靶向捕获扩增子建库测序步骤:第一轮多重长PCR,添加低浓度多重长PCR特异引物,靶向捕获含有高同源区段SNP位点的目标序列;第二轮多重PCR,取部分第一轮多重PCR产物作为模板添加到第二轮PCR体系中,并且采用荧光定量PCR法调整多重PCR引物浓度以保证多重PCR扩增的均一性;第三轮PCR,直接将第三轮PCR体系添加到第二轮多重PCR产物中,利用接头引物将所有PCR产物添加上测序接头。经过三轮PCR后,成功构建二代测序扩增子文库。
1.5 多重长PCR靶向捕获扩增子建库测序技术
1.5.1 第一轮PCR
PCR反应体系(10 μL)包括3.0 μL双蒸水,5 μL PCR Mix(2×Phanta®Flash Master Mix,南京诺唯赞生物科技股份有限公司,货号P520-01),1 μL混合特异引物(每条特异引物浓度为0.5 μmol/L)和1 μL DNA模板(质量浓度为20 ng/μL)。第一轮长PCR反应程序:98 ℃预变性120 s,98 ℃变性10 s,68 ℃退火10 s,72 ℃延伸90 s,2个循环;98 ℃变性10 s,66 ℃退火10 s,72 ℃延伸90 s,2个循环;98 ℃变性10 s,64 ℃退火10 s,72 ℃延伸90 s,2个循环;98 ℃变性10 s,62 ℃退火10 s,72 ℃延伸90 s,2个循环;98 ℃变性10 s,60 ℃退火10 s,72 ℃延伸90 s,20个循环。待PCR反应程序运行结束后,取下PCR反应体系进行磁珠纯化。第一轮长PCR产物磁珠纯化条件:取40 μL(2个相同的孔混在一起,样本与磁珠比例为1∶1.8)样本添加72 μL磁珠,混匀,静置2 min;置于磁力架30 min,弃上清;加180 μL质量分数为85%的乙醇,静置30 s,弃上清,挥发5 min;20 μL TE洗脱、吹打,脱离磁力架静置2 min;再次置于磁力架上吸上清15 μL ,收集在洁净的EP管中。对获得的第一轮长PCR纯化产物进行电泳检测,设置条件:各取5 μL收集的样本,5 k长marker 取2 μL,分别添加到质量分数为0.8%的琼脂糖凝胶的胶孔中进行电泳。
1.5.2 第二轮PCR均一性调整
将磁珠纯化后的多重长PCR产物作为模板,进行第二轮PCR。由于第一轮PCR的靶向捕获目标区段,第二轮PCR的靶向捕获目标区段得以避免同源基因的干扰。因此,第二轮PCR产物可用于构建扩增子文库,但需要通过定量PCR方法精确地调整多重PCR引物浓度,确保多重扩增子表达量一致。定量PCR反应体系(20 μL)包括10 μL的2×NovoStart®SYBR qPCR SuperMix (近岸蛋白质科技股份有限公司,货号:E096-01A),0.2 μL的ROX Reference Dye II ,1 μL F-Primer,1 μL R-Primer,2 μL DNA,5.8 μL ddH2O。PCR反应程序:94 ℃预变性60 s,95 ℃ 变性20 s,60 ℃退火45 s,72 ℃延伸45 s,35个循环。
1.5.3 第二轮PCR
PCR反应终体系(20 μL)包括4 μL双蒸水,2 μL第一轮PCR磁珠纯化产物作为模板,2.0 μL PCR master mix(近岸蛋白质科技股份有限公司,货号:E086-YSAA),2 μL Multiplex-Primer(每条特异引物0.5 μmol/L),10 μL Paraffin oil(货号:B500301-0500)。第二轮PCR反应程序如下:94 ℃预变性5 min,98 ℃ 变性15 s,60 ℃退火4 min,5个循环;98 ℃ 变性15 s,65 ℃退火15 s ,72 ℃延伸30 s,18个循环。待PCR反应程序运行结束后从PCR仪器中取出第二轮PCR反应产物进行磁珠纯化,磁珠纯化条件:取10 μL样本加10 μL磁珠(样本与磁珠比例为1∶1)混匀,静置2 min;置于磁力架30 min,弃上清;添加180 μL质量分数为85%的乙醇,静置30 s,弃上清,挥发5 min;添加20 μL TE溶液进行洗脱,吹打后脱离磁力架静置2 min;再次置于磁力架上并吸上清15 μL,保存于洁净的EP管中。
1.5.4 第三轮PCR
PCR反应体系(10 μL)包括6 μL双蒸水,2 μL第二轮PCR磁珠纯化产物(作为模板),4.0 μL AmpliMix(近岸蛋白质科技股份有限公司,货号:N231-N234A),4 μL Adapter(5 μmol/L),4 μL I5/I7,10 μL Paraffin oil(货号:B500301-0500)。第三轮PCR反应程序:94 ℃预变性5 min,98 ℃变性15 s,65 ℃退火30 s,72 ℃延伸30 s,18个循环;待PCR反应程序结束后从PCR仪器上取出第三轮PCR产物,将329个样本的PCR产物等量混合后振荡混匀。将混匀后的第三轮PCR产物利用TIANSeq Size Selection DNA Beads试剂盒(天根生化科技(北京)有限公司,货号:NG306)进行片段分选,再次纯化后获得的PCR纯化产物可直接用于后续测序反应,纯化条件同第1.5.3节。
1.6 illumina 平台测序纯化后的建库产物
由于PCR建库孔板数量限制,将329个样本分成4组(样本数分别为96、96、96、41)混合PCR产物,纯化后上机测序(由北京诺禾致源科技股份有限公司完成)。
1.7 二代测序数据分析
二代测序数据原始数据通常包含index序列、接头序列和特异序列3个部分。基于样本index序列的差异,利用FASTX-Toolkit软件比对index序列,根据index序列差异对原始数据中每个样本的数据进行区分;匹配到每个样本的数据利用Fastp软件(v 0.20.1)将接头序列去掉。运用序列比对软件BWA(v0.7.17)将去接头后的序列与SNP对应的参考序列进行比对,统计每个位点等位基因的reads数目,计算其等位基因比率。将二代测序数据分型结果以NCBI数据库中SNP基因频率为标准进行比对。
2 结果与分析
2.1 同源性评估结果
针对9个SNP位点的序列评估其200 bp和400 bp片段在基因组中的同源性情况,结果显示在SNP对应的400 bp和200 bp的片段中,rs1801272、rs28399454、rs28399468、rs4986893、rs5031016对应的高同源片段均为4;rs2279343、rs28399499、rs3745274、rs4244285对应的高同源片段均为2。SNP位置处于400 bp片段的中间位置的本地Blast评估结果见表3,SNP位置处于200 bp片段的中间位置的本地Blast评估结果见表4。

表3 SNP对应的400 bp序列同源性评估Table 3 Homology evaluation of 400 bp sequences corresponding to SNP

表4 SNP对应的200 bp序列同源性评估Table 4 Homology evaluation of 200 bp sequences corresponding to SNP
由表3、表4可知,表中同源性为4的5个SNP位点为rs1801272、rs2839945、rs28399468、rs4986893、rs5031016,同源性为2的4个SNP位点为rs2279343、rs28399499、rs3745274、rs4244285。9个SNP对应的400 与200 bp的同源片段个数均≥2。在试验中将其划分为高同源区段SNP位点,直接使用新方案对高同源区段SNP分型,无需尝试普通多重扩增子建库测序。避免在普通扩增子靶向测序的SNP分型中出现高同源区段SNP无法被检测或检测结果混乱的情况,从而为试验推进节约了大量的时间和试验材料。
2.2 测序结果
2.2.1 二代测序数据质控
结合329个样本,对9个高同源SNP位点进行分型,二代数据质控结果显示Q20>90%、Q30>85%,均符合illumina官方的二代测序数据质量标准。扩增子测序深度分布图如图3所示。
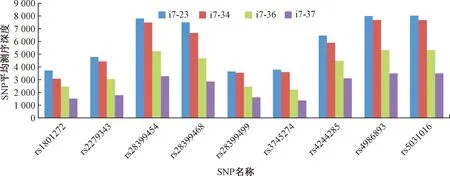
图3 Gatk3软件分析所得4组数据中的9个SNP位点测序深度Fig.3 Sequencing depth of 9 SNP loci in 4 groups of data analyzed by Gatk3 software
由图3可知,所有的SNP位点平均测序深度均大于1000,9个SNP的测序深度存在一定的偏差,但均能满足SNP位点的精确检出要求。
采用多重长PCR靶向捕获二代测序建库技术对高同源区段SNP分型时,利用Bio-Linux 8.04搭建的生信平台分析二代测序数据。由图4可知,检出的SNP测序深度总体在1 400和8 100之间波动,其中rs28399454、rs28399468、rs4244285、rs4986893、rs5031016测序深度偏高,rs1801272、rs2279343、rs28399499、rs3745274测序深度偏低。对比SNP同源性与对应SNP位点的测序深度发现,SNP对应的片段同源性大小与测序深度的相关性不显著。
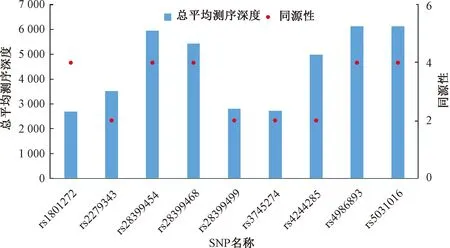
图4 4组数据中9个SNP位点的平均测序深度与同源性Fig.4 Average sequencing depth and homology of 9 SNP loci in 4 sets of data
2.2.2 二代测序扩增子等位基因比率
对329个样本的9个高同源区段的SNP进行检测,二代测序数据显示共获得2 928个SNP位点信息,分型结果如图5所示。

图5 329个样本的9个高同源SNP的等位基因分布情况Fig.5 Allele distribution of 9 highly homologous SNP in 329 samples
由图5可知,多重长 PCR 捕获二代测序技术检测 329 例人血液基因组高同源 SNP的9 个高同源 SNP 位点rs1801272、rs2279343、rs28399454、rs28399468、rs28399499、rs3745274、rs4244285、rs4986893、rs5031016的基因频率分别为A1.0、A/G0.74/0.26、C1.0、C1.0、T1.0、G/T0.826/0.174、G/A0.721/0.279、G/A0.953/0.047、A/G 0.988/0.012。二代测序数据显示共获得2 928个SNP位点信息,测序成功率为 98.885 6%。通过HWE法则计算所得SNP基因频率结果与PCR-LDR设置经验性阈值计算出的基因频率结果一致,表明SNP基因频率计算无误。
2.3 二代测序扩增子等位基因比率与NCBI中千人基因组数据库基因频率比对
多重长PCR靶向捕获扩增子建库测序对高同源区段SNP分型中的位点等位基因频率的分析结果与千人基因组数据库基因频率比对结果如表5所示。

表5 二代测序329个样本的高同源SNP分型结果与千人基因组数据库基因频率对比
由表5可知,千人基因组数据库中rs1801272、rs2279343、rs28399454、rs28399468、rs28399499、rs3745274、rs4244285、rs4986893、rs5031016基因频率依次为A/T1.0、A/G0.665/0.335、C/T1.0、C/A0.978/0.022、T/C1.0、G/T0.747/0.253、G/A0.708/0.292、G/A0.919/0.081、A/G 0.847/0.153。多重长PCR靶向捕获扩增子测序所得结果与数据库基因频率误差值均在 0.15 以内,二代测序检测结果可靠。通过该方案能够完成高同源区段SNP的准确、快速分型。
3 结 语
本研究通过在线Blast筛选高同源SNP片段,利用设计的长PCR引物对含9个SNP位点的目标序列进行高校靶向捕获。经两轮PCR扩增和测序,成功检测2 928个SNP,测序成功率高达98.89%。基因型频率分析中,将等位基因杂合子为15%~85%,分型结果与千人基因组数据库结果一致,误差不超过0.15。同时,研究结果还表明同源性高低不是影响SNP位点测序深度的因素。
因此,本研究结合本地Blast评估同源性,采用Oligo7.6和Primer3-py软件进行引物互作分析,有效提高多重PCR的成功率。建立了一个专注于高同源区段SNP分型的多重长PCR靶向捕获扩增子建库测序方案。该方案提出生信前处理、多重引物互作和多重长PCR靶向捕获测序的新流程,成功解决了靶向高通量测序SNP分型中的脱靶问题,大幅节约了试验时间和材料成本,为二代测序SNP分型做了重要的补充。
