金合金中Ce和Lu晶界偏聚的第一性原理研究
2024-02-28刘浩松谢耀平周文艳胡丽娟姚美意
刘浩松 谢耀平 周文艳 胡丽娟 姚美意
( 1.上海大学 材料科学与工程学院,上海 200072; 2.贵研铂业股份有限公司 稀贵金属综合利用新技术
国家重点实验室,云南 昆明 650106)
元素的晶界偏聚通常会引起材料的强度、韧性、耐蚀性等性能的变化[1-4]。因此,钢[5-7]、锆合金[8]、铝合金[9]、镁合金[10]、氧化物[11]等材料中溶质元素的晶界偏聚与力学性能之间的关系得到了广泛研究[12-13]。 Araki等[14]研究发现,晶界碳元素的偏聚会阻碍晶界附近的位错滑移,导致晶界剪切应力增加,从而提高铁素体钢的强度。Dai等[15]采用蒙特卡罗和分子动力学相结合的方法研究了钨在ZrB2晶界的偏聚,发现钨有强烈偏聚倾向,并诱导晶界结构重构,进而导致晶界强度显著提高。Masuda等[16]采用原子探针和透射电子显微镜,从晶界特性、晶粒尺寸和位错密度等方面阐明了晶界偏聚对A2024铝合金拉伸强度的提高具有重要作用。Nie等[17]研究镁合金的偏聚行为发现,溶质原子在孪晶界的有序偏聚产生了钉扎效应,导致退火强化合金而不是弱化合金,这为指导新的合金成分和热加工方法提供了可能。Xu等[18]利用第一性原理研究了Cu对锆合金氧化物中单斜氧化锆晶界抗拉强度的影响,发现Cu原子的晶界偏聚提高了晶界的抗拉强度,从而提高了锆合金的耐蚀性。
随着芯片尺寸的减小及混合集成技术的发展,半导体器件的集成度进一步提高,这对芯片封装技术也提出了更高的要求[19-20]。目前多芯片系统中最常用的封装方法是引线键合技术,键合丝在芯片与引线框架之间进行电气互连[21],金键合丝由于其惰性和优异的循环性能,是半导体器件引线键合中使用最多的丝材[22]。为了确保引线键合过程中良好的焊接性能,需要直径更细且强度更高的金键合丝[23]。向金键合丝中添加合金元素以获得优异的力学性能是满足芯片封装新要求的有效方法,而合金元素的晶界偏聚是金合金研究中不可避免的问题。目前,国内对添加合金元素改善金键合丝性能的研究虽然取得了一定进展,但对合金元素的强化机制仍不清楚[24-25]。而国外,如日本制造出了世界一流的金键合丝并已成熟应用[26]。因此,为了推进国内金键合丝的技术升级,需要深入地研究合金元素对金键合丝的强化机制。
昆明贵金属研究所试验研究发现[27],以高纯金(Au≥99.999%)与纯度大于99.95%的微量元素Ce和Lu为原材料进行合金化处理,Ce元素的晶粒细化效果显著,而Lu元素只有轻微的晶粒细化作用。为了研究两者细化晶粒效果差异的原因,深入理解合金元素在金中的偏聚行为及影响机制,本文采用第一性原理计算研究了Ce和Lu在纯金晶界的偏聚能和偏聚倾向,并结合原子结构、电子结构分析揭示了Ce和Lu在纯金晶界的偏聚机制。
1 计算方法与模型构建
1.1 第一性原理计算方法
本文利用基于密度泛函理论(density functional theory, DFT)[28]的第一性原理计算方法(first- principles method),对Ce和Lu在纯金晶界附近的偏聚能进行计算。计算中离子与价电子之间的相互作用由投影缀加波(projector-augmented wave, PAW)描述[29-30],选择广义梯度近似(generalized gradient approximation, GGA)[31]下的PBE(Perdew-Burke-Ernzerhof)泛函[32]作为电子间的交换关联泛函,k点网格使用Monkhorst-Pack方法进行采样[33]。本文涉及计算k点网格均设置为5×5×1,平面波展开的截断能设定为540 eV,对参数进行测试,验证了截断能和k点网格密度已经达到了能量收敛标准为0.01 eV/atom的精度要求。
1.2 模型构建
本文使用Slab模型模拟金合金结构,在保证计算精度的同时节省计算成本,使用相对较小的∑5{210}/[001] 29°超胞模型进行计算。
如图1所示,构建了纯金理想晶格和晶界Slab模型,图1(a)中理想晶格模型由4个取向相同的{210}晶面原子层周期组成,每个周期有10层原子,共40个原子规则排列。图1(b)中纯金晶界模型的周期数、原子数和理想晶格模型相同,原子周期沿晶界对称。因此,本文以晶界附近目标周期的10个位点作为偏聚位点进行研究。两个模型具有对应的结构和大小,因此可以抵消由超胞的结构和大小引起的能量误差,保证能量都来源于合金元素在晶界附近的偏聚。

图1 纯金理想晶格(a)和晶界(b)Slab模型(黄色原子表示Au原子,目标周期用数字1~10标记,蓝色虚线表示晶界;为了方便作图,(a)中在另一个周期与1号原子相对应的原子被标为11,(b)中与2号原子相对于晶界对称的原子被标为-2)
2 计算结果与讨论
2.1 元素偏聚行为
Ce和Lu元素在晶界的偏聚能计算公式为:
(1)

根据式(1),通过第一性原理计算出Ce和Lu在纯金晶界附近1~10号位点的偏聚能,结果如图2所示。
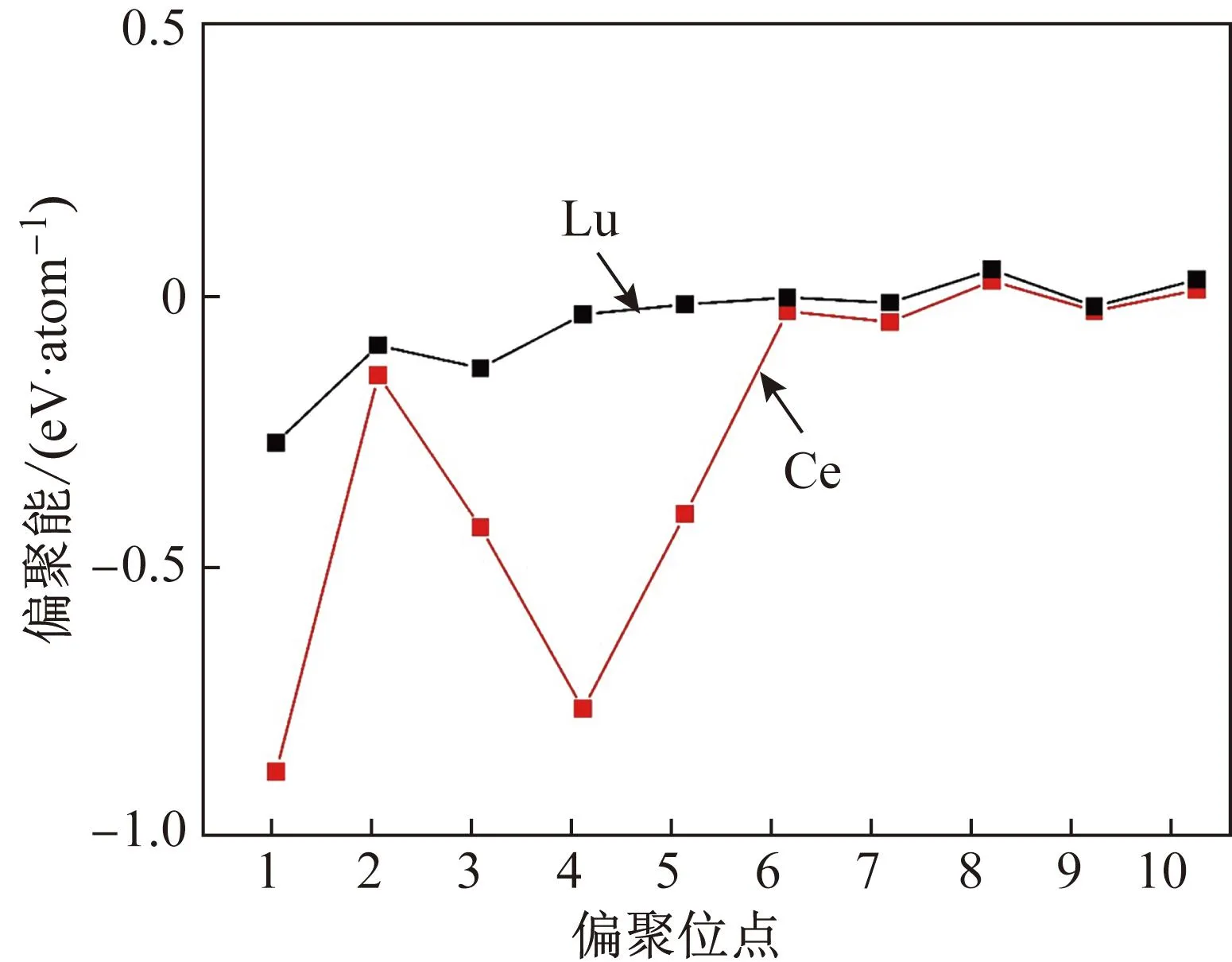
图2 Ce和Lu在纯金晶界附近1~10号位点的偏聚能Fig.2 Segregation energy of Ce and Lu at sites No.1 to 10 near the grain boundary in pure Au
从图2可以发现,Ce和Lu的偏聚能都在-1.0~0.5 eV范围内波动。偏聚能为负数表示偏聚是自然发生的放热反应,而正数表示不能自然发生的吸热反应。分析发现,1号位点Ce和Lu的偏聚能均最小,分别为-0.89和-0.28 eV;随着远离晶界,Ce和Lu的偏聚能都逐渐趋于零。这是因为晶界附近的原子结构与理想晶格差异较大,偏聚后体系能量变化更大,偏聚容易发生;反之,远离晶界,原子结构更接近理想晶格,偏聚后体系能量变化较小,偏聚难以发生。偏聚能低于-0.5 eV定义为强偏聚体系[34],Ce为强偏聚元素,Lu为弱偏聚元素。
为了更准确地评估Ce和Lu的偏聚行为,使用McLean公式[35]进一步计算偏聚倾向:
(2)
式中:CGB表示合金元素在晶界附近的偏聚倾向;Cbulk表示合金元素在纯金理想晶格内的质量分数,本文为0.01%;ΔEseg表示合金元素的偏聚能,T表示偏聚过程的时效温度,本文为500 ℃。计算得出Ce在纯金晶界的最大偏聚倾向为99%,Lu为1%。
2.2 原子结构分析
为了进一步研究Ce和Lu在纯金晶界偏聚的物理成因,分析了Ce和Lu的晶界偏聚与原子结构之间的关系。基于pure Au、Ce@Au和Lu@Au 3种体系的理想晶格和晶界模型(Ce、Lu都位于1号位点),统计了图1中1~10号位点原子近邻最大Lmax和最小键长Lmin,结果如表1、表2所示。
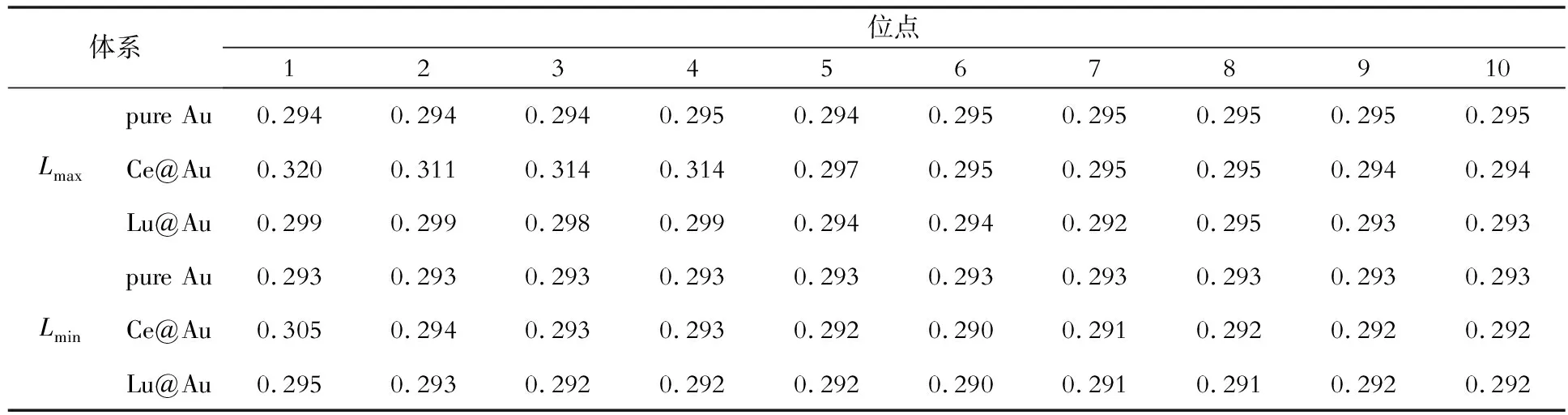
表1 理想晶格结构中Ce和Lu位于1号位点时1~10号位点原子的最大、最小键长(Lmax和Lmin )Table 1 Maximum and minimum bond lengths (Lmax and Lmin) of atoms at sites No.1 to 10 when Ce and Lu are located at site No.1 in the ideal crystal structure nm
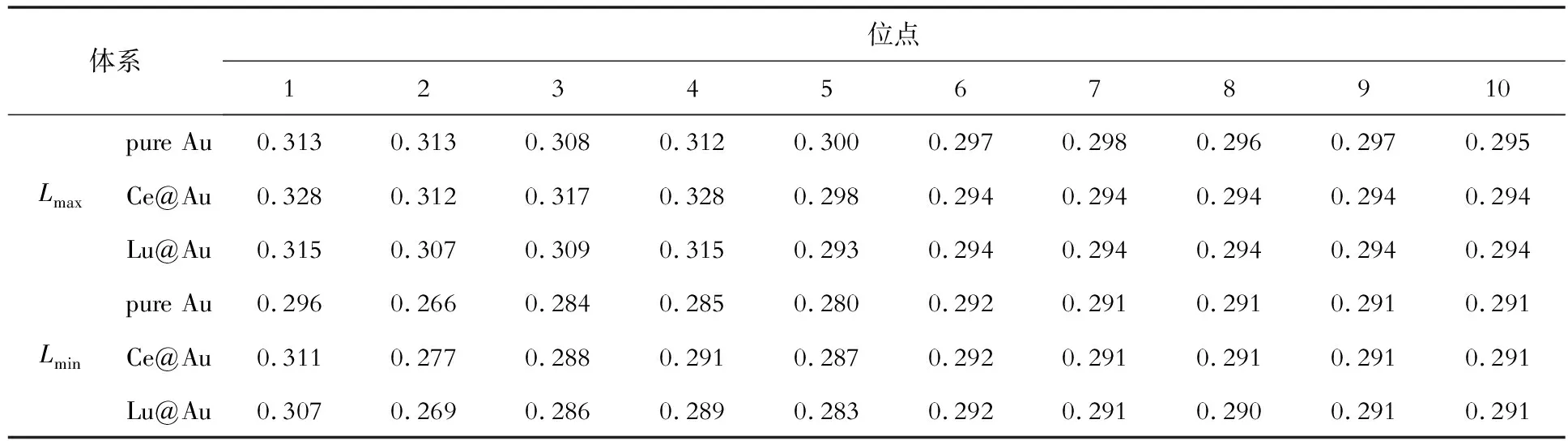
表2 晶界结构中Ce和Lu位于1号位点时1~10号位点原子的最大、最小键长(Lmax和Lmin)Table 2 Maximum and minimum bond lengths (Lmax and Lmin) of atoms at sites No.1 to 10 when Ce and Lu are located at site No.1 in the grain boundary structure nm
由表1可知,纯金理想晶格结构中,1~10号原子键长范围为0.293~0.295 nm。Ce和Lu位于1号位点时,1~5号原子的键长范围分别为0.292~0.320 nm和0.292~0.299 nm,相较于纯金理想晶格明显增大;6~10号原子的键长范围都为0.290~0.295 nm,相较于纯金理想晶格变化较小。理想晶格中Ce和Lu原子附近1~5号原子引起了明显晶格畸变,其中Ce引起的畸变更大,体系能量变化更大;随着远离Ce和Lu所在1号位点,畸变减小,6~10号原子键长接近理想晶格。
由表2可知,纯金晶界结构1~5号原子键长范围为0.266~0.313 nm,6~10号原子键长范围为0.291~0.298 nm。Ce和Lu位于1号位点时,1~5号原子的键长范围分别为0.277~0.328 nm和0.269~0.315 nm,相较于纯金晶界结构明显增大;6~10号原子的键长范围分别为0.291~0.294 nm和0.290~0.294 nm,相较于纯金晶界变化较小。Ce和Lu在晶界同样会引起明显的晶格畸变,Ce引起的畸变依然大于Lu。
对比表1、表2数据可知,3种体系晶界的1~5号原子键长均明显大于理想晶格的,符合晶界附近的原子结构与理想晶格差异较大的规律。理想晶格的原子结构比较致密,而晶界附近的原子结构比较疏松,Ce和Lu在晶界附近引起的晶格畸变小于理想晶格内部,这是晶界附近偏聚能为负数的原因;而随着远离晶界,原子结构与理想晶格逐渐趋于一致,偏聚能接近于零。同时,Ce在晶界偏聚后的键长大于Lu,表明Ce在纯金晶界结构中会诱发更大的晶格畸变,导致更大的能量变化,Au-Ce键要强于Au-Lu键,这与Ce在晶界的偏聚倾向更大是一致的。
2.3 电子结构分析
为了从电子结构角度分析Ce和Lu的偏聚机制,计算了3种体系理想晶格和晶界结构的差分电荷密度图,如图3所示。由图3可知,Au、Ce、Lu靠近原子核区域的电子向远离原子核区域转移,形成巡游电子,这是金属晶体的典型特征。
分析图3(a)理想晶格结构发现,相较于Au原子的电荷变化,Ce@Au体系的Ce原子以及附近的Au原子发生了明显的电荷转移;Lu@Au体系的Lu原子也发生了电荷转移,但程度明显小于Ce。分析图3(b)晶界结构发现,3种体系都出现了明显的电荷转移。同时,晶界结构中靠近Ce原子核位置为明显失电子区域,比理想晶格结构中Ce原子失去的电子更多,表明Ce在晶界有更强的金属性。而晶界结构中Lu原子的电荷行为明显不一样,Lu原子失电子并没有比理想晶格中Lu原子失电子更多,Lu在晶界的金属键没有增强。
对比Ce和Lu在理想晶格和晶界结构中的电荷状态,发现Ce的失电子效应明显强于Lu。根据电负性的定义可知,电负性是将电子引向自身的能力[36],而Ce吸引电子的能力更弱,电负性更小。鲍林(Pauling)标度是目前应用最广泛的电负性标度,根据此标度可知,Ce和Lu的原子电负性分别为1.12和1.27[37]。差分电荷密度和Pauling标度均表明,Ce电负性明显小于Lu,而电负性更小的金属元素更容易失电子,因此Au-Ce金属键比Au-Lu更强,Ce更容易在Au晶界发生偏聚。Ce在晶界结构中的金属键比在理想晶格中的金属键更强,贡献了更大的偏聚能;而Lu在晶界结构中的金属键没有得到加强,这也是Ce的偏聚能小于Lu的重要原因。

图3 理想晶格结构中穿过1、2、11号位点原子(a)、晶界结构中穿过1、2、-2号位点原子并垂直于晶界平面(b)的平面差分电荷密度图(图中数字表示原子位点,键长距离用实线及数值标出,nm;平面Δρ为-0.3~0.8 e/bohr3,轮廓线间隔为0.03 e/bohr3;白色和浅灰色区域表示失电子,深灰色和黑色区域表示得电子)
计算态密度可以获得更详细的电荷状态,图4为3种体系理想晶格和晶界结构的总态密度(total density of states, TDOS)和分波态密度(partial density of states, PDOS)。分析图4(a,c)TDOS图可以发现,Ce@Au体系在费米面附近的态密度明显更高,而Lu@Au体系在费米面附近的态密度很低;从图4(b,d)PDOS图可以发现,Ce@Au体系费米面附近的态密度更高。其原因是理想晶格和晶界结构中的Ce原子在费米面附近的态密度远高于Au原子,而Lu原子在费米面附近的态密度低于1个Au原子。态密度结果表明,Ce在理想晶格和晶界结构费米面附近的态密度均更高,化学性质更活泼,更容易失电子,形成更强的金属键,这与差分电荷密度分析的结果一致。
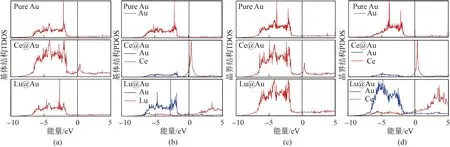
图4 理想晶格和晶界结构的(a,c)总态密度和(b,d)分波态密度(费米面在0 eV位置)Fig.4 Total density of states (a,c) and partial density of states (b,d) of ideal crystal and grain boundary structures (Fermi surface being at 0 eV )
2.4 讨论
由上文分析可知,Ce比Lu引起的晶格膨胀更大,这可以从原子半径角度进行解释。Ce和Lu在纯金中以金属键的形式存在,所以主要关注金属半径(r),即相邻两个原子互相接触时两核间距离的一半[38]。因此,对Au、Ce和Lu原子的金属半径进行比较,发现不同文献中原子金属半径的取值不同。典型的如剑桥晶体结构数据库(cambridge structural database, CSD)中,基于X射线和中子衍射分析的数据[39],Wells利用X射线晶体学测量的数据[40],Mann基于狄拉克-哈特里-福克(Dirac-Hartree-Fock)公式计算的镧系元素的原子半径[41],如表3所示。分析3组数据可以发现,3种金属原子半径的试验值和计算值虽然存在差异,但大小规律一致,即rCe>rLu>rAu,表明在不同测定体系中Ce的金属半径始终大于Lu,从而部分解释了Ce在理想晶格和晶界结构中均引起更大晶格畸变的原因。

表3 不同文献中Au、Ce和Lu的金属半径Table 3 Metallic radius of Au, Ce and Lu from different literatures nm
Ito等[6]认为,原子结构中的原子尺寸差是影响晶界偏聚的重要因素,通常弹性能对晶界偏聚能的贡献最大,弹性能与溶质元素和主体金属元素之间的原子尺寸差成正比,尺寸差越大,弹性能的贡献越大,偏聚能越小。对比计算结果发现,Au和Ce的原子尺寸差更大,引起更大的晶格畸变,弹性能贡献更大,偏聚能更小,这与lto等的结论是一致的。另外,计算结果显示,Ce在晶界的金属键增强也是偏聚能更小的重要原因之一。
本文从原子结构和电子结构两个角度揭示了Au合金中Ce和Lu偏聚倾向出现明显差异的原因。目前,Au合金体系中晶界偏聚的试验结果尚未见报道。而昆明贵金属研究所的试验结果表明[27],Ce元素在Au合金中的晶粒细化效果明显大于Lu元素,由于试验添加的Ce和Lu质量分数均为0.01%,含量很低,形成固溶体或者析出相的概率较小,最可能的是合金元素的晶界偏聚,与晶界产生相互作用,从而抑制晶界迁移,细化晶粒,这与本文计算得到的Ce和Lu的偏聚倾向结果一致。
3 结论
(1)在纯金晶界附近1~10号位点,Ce和Lu均在1号位点偏聚最强,偏聚能分别为-0.89和-0.28 eV,最大偏聚倾向分别为99%和1%,Ce为强偏聚元素,Lu为弱偏聚元素。
(2)Ce和Lu都处于1号位点时,讨论3种体系理想晶格和晶界结构的1~10号原子的化学键长,发现Ce相较于Lu,在理想晶格和晶界结构中均引起了更大的晶格畸变,导致更大的能量变化,Au-Ce键强于Au-Lu键,这与Ce在晶界偏聚的倾向更大一致。
(3)分析理想晶格和晶界结构的差分电荷密度,发现Ce更容易失电子,Ce的电负性小于Lu,拥有更多的活跃电子,Au-Ce键比Au-Lu键强;Ce在晶界结构中的金属键比在理想晶格结构中的更强,而Lu在晶界结构中的金属键没有增强,是造成Ce在晶界偏聚能低的重要原因。
