HPLC法测定溴芬酸钠滴眼液中聚合物的含量
2024-02-27吴永芹
吴永芹 薛 雯 孟 龙 王 军 韩 勇 张 莹
1.山东省济宁市食品药品检验检测研究院,山东济宁 272073;2.山东省食品药品审评查验中心,山东济南 250014
溴芬酸钠,即[2-氨基-3-(4-溴苯甲酰基)苯基]乙酸钠,其制剂0.1%溴芬酸钠滴眼液于2000年在日本上市,于2015年在国内批准生产上市。0.1%溴芬酸钠滴眼液临床应用较广,主要用于外眼部及前眼部的炎症的治疗,如眼部手术后的炎症、结膜炎等症状,其作用强度是其他非甾体抗炎药的十倍,临床效果显著[1-6]。配方中的增稠剂聚维酮K30在储存过程中与溴芬酸钠发生聚合反应,生成聚合物杂质U-Ⅱ,会降低主药含量,同时使药液黏性下降,疗效降低,聚合物杂质U-Ⅱ的产生同时增加了使用过程中的不良反应发生风险。本研究建立HPLC对溴芬酸钠滴眼液中产生的聚合物的含量进行测定,为控制其药品质量提供方法依据。
1 仪器与试药
LC-2030C型高效液相色谱仪(DAD检测器)(日本岛津);流动相吸附柱(4.6 mm×50 mm)(中谱科技),色谱柱:Waters C18(4.6 mm×250 mm,5 μm);TE214S型电子天平、BS21S型电子天平[赛多利斯科学院仪器(北京)有限公司];FE28酸度计(梅特勒-托利多仪器有限公司)、磷酸氢二钾(天津市光复精细化工研究所,优级纯);乙腈(天津四友,HPLC级);磷酸(阿拉丁,HPLC级);超纯水(实验室自制,电阻率18.2 MΩ);溴芬酸钠对照品(LGC,W1008660);聚维酮对照品(中检院,批号:190070-201501),溴 芬 酸 钠 滴 眼 液[批 号:0A0021M10、0A0011M10(A厂 家),1907033101、1907053101、1912073102、2001013101、2001033101、2006043101、2007063101、2007083101(B厂家),P432(C厂家)]。
2 方法与结果
2.1 色谱条件
流动相A为0.05 mol/L磷酸氢二钾溶液(pH 4.5)-乙腈(75∶25),流动相B为乙腈;梯度洗脱:0~5 min(0%B),5~10 min(0%→28%B),10~30 min(28%B),30~30.1 min(28%→0%B),30.1~60 min(0%B);柱温30℃,流速1.0 ml/min,检测波长400 nm,进样量20 μl。
2.2 溶液的制备
2.2.1 对照品溶液的制备 精密称取溴芬酸钠对照品10.01 mg,置100 ml棕色量瓶中,加入30 ml流动相A,振摇使对照品溶解,用流动相A稀释至刻度,摇匀,作为对照品溶液①,精密量取1 ml,置100 ml棕色量瓶中,用流动相A稀释至刻度,摇匀,即得。
2.2.2 供试品溶液的制备 精密量取溴芬酸钠滴眼液2 ml,置10 ml棕色量瓶中,加入流动相A振摇并稀释至刻度,摇匀,即得。
2.2.3 聚维酮K30阴性对照溶液的制备 制备不含聚维酮K30的阴性供试品,精密量取2 ml,置10 ml棕色量瓶中,加流动相A振摇并稀释至刻度,摇匀,即得。
2.2.4 聚维酮K30对照溶液的制备 取聚维酮K30原料,加流动相A制成浓度为2 mg/ml的溶液。精密量取2 ml,置10 ml棕色量瓶中,加流动相A振摇并稀释至刻度,摇匀,即得。
2.3 专属性试验考察
精密吸取上述溶液各20 μl进样测定,见图1,结果表明,聚合物峰与相邻色谱峰分离良好,出峰时间约为10 min,阴性对照无干扰,在400 nm波长处聚维酮K30无吸收,对聚合物的测定无干扰。

图1 专属性试验色谱图
2.4 线性关系考察
分别精密量取2.2.1下不同体积的对照品溶液①,置50 ml棕色量瓶中,用流动相A稀释至刻度,摇匀,制备得到浓度为0.1、0.2、0.5、1.0、5.0、10.0 μg/ml的系列标准溶液,精密吸取20 μl,进样测定,以浓度C(μg/ml)为横坐标,峰面积A(μv·ms)为纵坐标作图,得到其回归方程为A=11463.95C-130.90,r=0.9995,结 果 表 明 在 进 样 量 为20 μl时,在0.1~10.0 μg/ml浓度范围内,线性关系良好。见图2。
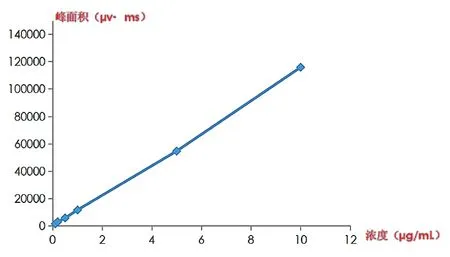
图2 线性关系曲线图
2.5 精密度试验
精密量取对照品溶液(浓度为1 μg/ml),进样6次,峰面积(μv·ms)分别为14 484、14 433、14 459、14 535、14 499、14 382,RSD为0.38%。取 供试品溶液,重复进样6次,峰面积(μv·ms)分别为14 753、14 684、14 293、14 574、14 154、14 982,RSD为2.1%。试验结果表明,仪器的精密度良好。
2.6 稳定性试验
考察溶液24 h稳定性,峰面积(μv·ms)分别为18 468(0 h)、17 463(2 h)、18 132(4 h)、18 088(8 h)、16 268(12 h)、12 543(24 h),结果样品溶液在8 h内稳定,8 h内RSD为2.9%。
2.7 重复性试验
取溴芬酸钠滴眼液,按照供试品溶液制备方法制备供试品溶液,共6份,进样测定,峰面积(μv·ms)分别为13 489、13 198、13 734、13 216、13 324、13 148,以峰面积计算,RSD为1.7%,试验结果表明,样品的重复性试验良好。
2.8 检出限和定量限
取溴芬酸钠对照品溶液,逐步稀释,按照信噪比S/N=3确定检出限溶液的浓度,检出限溶液浓度为0.02 μg/ml,按照信噪比S/N=10确定定量限溶液的浓度,定量限溶液浓度为0.05 μg/ml。将供试品溶液稀释5倍,精密吸取20 μl,进样测定,色谱图中聚合物峰的信噪比S/N=10,计算聚合物含量,定量限浓度相当于溴芬酸钠标示量的0.03%。
2.9 测定结果
按照建立的测定方法,测定三个厂家的11批样品,聚合物含量按照溴芬酸钠标示量计算,结果范围为0.1%~1.5%,分别按照各自现行法定标准进行测定,结果范围为0.1%~3.2%,聚合物测定结果见表1。
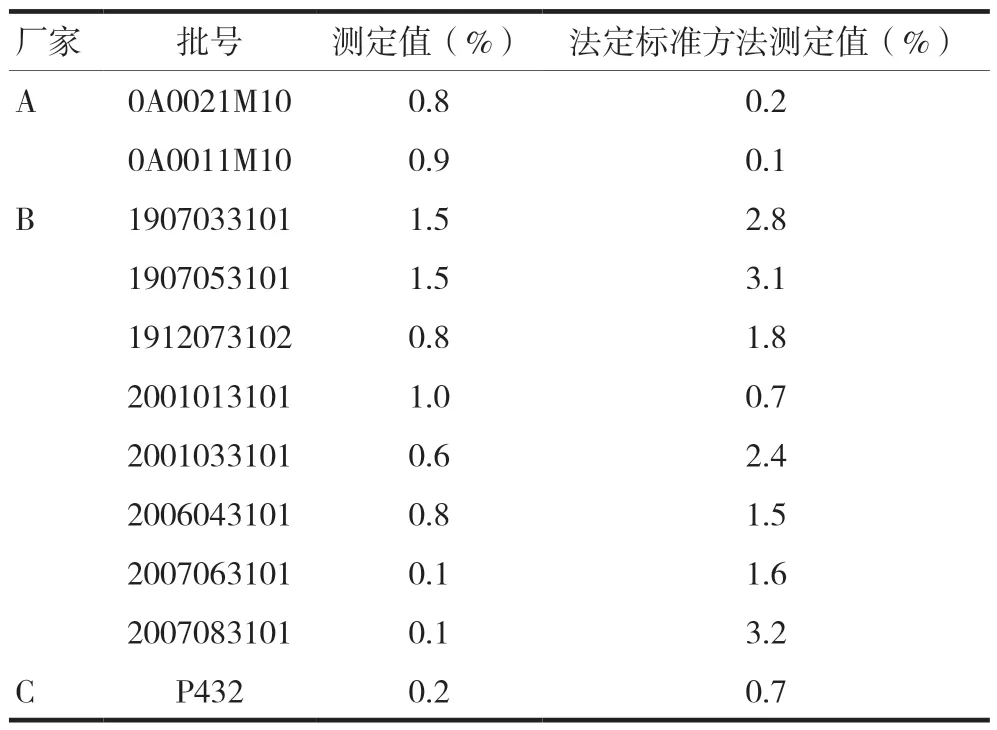
表1 聚合物测定结果(n=3)
3 讨论
本研究考察了不同pH值的0.1%三乙胺-乙腈体系、不同pH值的0.0025 mol/L庚烷磺酸钠-乙腈体系、不同pH值的0.05 mol/L磷酸氢二钾溶液-乙腈体系;考察了C8色谱柱、C18色谱柱、凝胶色谱柱G-10、TSKgel SuperMultiporePW-M尺寸排阻色谱柱;考察了蒸发光散射检测器、紫外检测器、示差折光检测器。当采用C18色谱柱,紫外检测器,以0.05 mol/L磷酸氢二钾溶液-乙腈体系为流动相时,聚合物与其他组分分离效果最好,可以进行准确定量。由于聚维酮K30为大分子物质,在C18色谱柱上出峰峰型较宽,且在色谱柱有残留,需提前进行色谱柱的饱和[7-17]。
聚合物是溴芬酸钠与聚维酮K30在药品的储存期间聚合产生的,无法分离纯化得到聚合物杂质纯品,市面上无聚合物杂质对照品出售,无法用对照品进行定量。现行国内外标准中聚合物的计算方式,采用样品峰面积直接扣除空白溶液中聚维酮K30峰面积,取峰面积差值,按外标法进行计算。聚维酮K30对照品的称样量或者峰面积值大小直接影响计算结果,甚至会出现负值,测定结果与聚维酮K30投料的多少和聚维酮K30对照品取样量的多少直接相关,存在监控风险。经比较分析溴芬酸钠对照品和聚维酮K30原料的紫外吸收光谱图,溴芬酸钠在400 nm波长处有较大的吸收,而聚维酮K30在400 nm波长处无紫外线吸收,不影响聚合物的定量,故检测波长选择为400 nm。
现行标准方法的供试品色谱图中聚合物峰型极不规则,基线不平,且计算方式不合理,含量测定的结果不准确,测定结果值差异较大。本研究选择在无干扰的400 nm波长处采集信号,以外标法直接计算含量,与现行标准方法比较,结果更准确可靠。但由于各个批次供试品聚合物的形成具有不确定性,结果差异也较大。
本研究建立了溴芬酸钠滴眼液中聚合物的含量测定方法,方法简便、快速,可用于控制溴芬酸钠滴眼液聚合物的含量,也为其质量控制方法的研究提供了参考。
