UHPLC-MS/MS测定人血浆中的阿普斯特浓度及生物等效性研究
2024-02-26张迅杰贺美莲杭宝建石峰巩丽萍张乃斌咸瑞卿
张迅杰,贺美莲,杭宝建,石峰,巩丽萍,张乃斌,咸瑞卿,2
(1.山东省食品药品检验研究院,国家药品监督管理局仿制药研究与评价重点实验室,山东省仿制药一致性评价工程技术研究中心,产业技术基础公共服务平台,山东 济南 250101;2.山东大学药学院,山东 济南 250012)
银屑病是一种常见的免疫相关的慢性复发性炎症性皮肤病[1],是一种多基因遗传和环境相互作用下,主要由细胞免疫异常介导的慢性炎症性增殖性皮肤病[2]。在世界范围内有1.25亿人患病,中国银屑病患者达600万以上[3]。银屑病关节炎[4]是一种与银屑病相关的炎性关节病。白塞病[5]是一种复杂的、原因不明的、多系统受累的慢性自身免疫病。
阿普斯特是一种口服的环单磷酸腺苷(cAMP)特异性磷酸二酯酶-4(PDE4)小分子抑制剂。PDE4的抑制能够升高细胞内的cAMP水平,PDE4降解cAMP可导致免疫细胞激活和促炎细胞因子释放[6-7]。因此抑制PDE4可能能够降低PDE4介导的炎症反应。阿普斯特片,2014年3月21日在美国获批上市销售,批准的适应证为活动性银屑病关节炎[8],适用光疗或者系统治疗的中度至重度斑块状银屑病和与白塞病相关的口腔溃疡。2014 年 9 月,FDA 批准阿普斯特用于治疗中重度斑块状银屑病[9]。由于其有希望的治疗结果和安全性,该药物分子在皮肤病学中变得非常流行[10]。目前国内有关测定人体内阿普斯特血药浓度的方法尚未研究,本研究首次建立了一种测定血浆中阿普斯特浓度的超高效液相色谱-串联质谱(UPLC-MS/MS)方法,该分析方法分析速度快,选择性好,准确度高,灵敏度高,基质效应小等优点。此外,对该分析方法进行了完整验证,以确保数据测定的准确性。利用此方法进行阿普斯特在中国健康受试者空腹及餐后状态下体内的药动学及人体生物等效性研究,为阿普斯特相关制剂一致性评价提供依据。
1 实验部分
1.1 仪器与试剂AB SCIEX Triple Quad 5500液相色谱-质谱联用系统(美国AB SCIEX公司);3K15台式高速冷冻离心机(德国Sigma公司);Allegra X-15R台式高速冷冻离心机(96孔板,美国贝克曼库尔特公司);Milli-Q Advantage A10超纯水器(美国Millipore公司)。数据处理软件:Analyst 1.6.3(美国AB SCIEX公司);Watson LIMS 7.5 SPS1(美国赛默飞世尔公司)。
阿普斯特对照品(纯度99.7%,北京曼哈格生物科技有限公司);阿普斯特-d5对照品(化学纯度98.7%,同位素内标纯度99.5%,北京曼哈格生物科技有限公司);甲醇、乙腈(色谱纯,德国默克公司);甲酸(色谱纯,美国ACS恩科化学);N,N-二甲基甲酰胺[色谱纯,赛默飞世尔科技(中国)科技有限公司]。受试制剂:阿普斯特片(规格:30 mg,批号:02007001);参比制剂:阿普斯特片(规格:30 mg,批号:H02366A,Celgene International Sarl 生产)。
1.2 分析条件
1.2.1 UHPLC条件Phenomenex Kinetex C18色谱柱(2.1 mm×50 mm,2.6 μm);柱温为40 ℃ ;流动相A相为0.1%甲酸水,B相为0.1%甲酸乙腈;采用梯度洗脱,洗脱程序为:0~0.5 min,流动相A的体积百分含量保持63%,流动相B的体积含量保持37%;0.5~1.3 min,流动相A的体积百分含量从63%下降到0.0%,流动相B的体积百分含量从37%上升到100%;1.3~2.4 min,流动相A的体积百分含量保持0.0%,流动相B的体积含量保持100%;2.4~2.41 min,流动相A的体积百分含量从0.0%上升到63%,流动相B的体积百分含量从100%下降到37%;2.41~3.0 min,流动相A的体积百分含量保持63%,流动相B的体积含量保持37%。流速为0.45 mL·min-1;进样量为5 μL。
1.2.2 MS条件采用电喷雾离子源(ESI)负离子模式;离子源温度参数设置为500 ℃ ;检测模式采用多反应监测(MRM)模式;离子化电压参数设置为5 500 V ;入口电压(EP)参数设置为10 V;碰撞室出口电压(CXP)参数设置为13 V;喷雾气(Gas1):50;辅助加热气(Gas2):55;气帘气:35。监测离子对:阿普斯特:461.2/257.1,碰撞能量(CE):14 V,去簇电压(DP):190 V;阿普斯特-d5:466.1/262.3,CE:14 V,DP:190 V。
1.3 对照品溶液制备
1.3.1 分析物(阿普斯特)工作溶液的配制精密称取阿普斯特适量,用N,N-二甲基甲酰胺(DMF)配制成质量浓度为 1.0 mg·mL-1的阿普斯特储备液Ⅰ,精密量取1 mL阿普斯特储备液Ⅰ适量置10 mL容量瓶中,加50%乙腈水溶液至刻度,使质量浓度为100 μg·mL-1阿普斯特工作溶液Ⅰ,避光保存于-20 ℃ 冰箱中。
1.3.2 内标(阿普斯特-d5)溶液的配制精密称取阿普斯特-d5适量,用N,N-二甲基甲酰胺(DMF)配制成质量浓度为2.0 mg·mL-1的阿普斯特-d5储备液Ⅰ,精密移取阿普斯特-d5储备液Ⅰ适量,用50%乙腈水溶液稀释至质量浓度为50.0 ng·mL-1阿普斯特-d5工作溶液Ⅰ,精密移取阿普斯特-d5工作溶液Ⅰ适量,用乙腈溶液稀释至质量浓度为20.0 ng·mL-1阿普斯特-d5工作溶液,避光保存于-20 ℃ 冰箱中。
1.4 样品预处理取2 mL深孔96孔板,每孔加100 μL血浆样品,再加质量浓度为20.0 ng·mL-1内标工作溶液(内标工作溶液稀释剂为乙腈溶液)300 μL,涡旋混合5 min,置4 ℃台式高速冷冻离心机(96孔板)中离心10 min(4 750 r·min-1),每孔取100 μL上清液,转移至另一2 mL深孔96孔板,向新的96孔板中加10%乙腈水溶液100 μL稀释,涡旋混合5 min,待测。
1.5 试验设计本次临床研究筛选24例健康成年受试者,所有受试对象均自愿参加本次临床试验,且均已签署书面知情同意书并理解研究程序,能按试验方案要求完成本研究项目。
本次临床研究采用单中心、单次给药、随机、开放、双周期、双制剂、双交叉试验设计,进行空腹及餐后两种状态下的制剂间生物等效性研究。空腹组12例受试者,餐后组12例受试者,空腹组和餐后组均随机分为两组,每组6人,按照给药顺序表分别给予受试制剂和参比制剂。所有受试者给药方法:空腹或者开始高脂餐后30 min口服,240 mL温水服用,给药剂量为30 mg。第Ⅰ周期6例受试者口服阿普斯特片30 mg(受试制剂),另6例受试者餐后口服阿普斯特片30 mg(参比制剂),7 d后进行第Ⅱ周期交叉给药研究。空腹/餐后试验: 在给药 0 h (给药前 1 h内)和 0.25、0.5、0.75、1、1.5、2、2.5、3、3.5、4、4.5、5、8、10、12、24、36 h,共 18 个时间点采集静脉血4 mL,空腹/餐后采集的静脉血均置EDTA-K2抗凝真空采血管中, 室温放置, 离心 (条件设定为 2 000 g、4 ℃、10 min),全血样品于采集完后 1 h内放入离心机。待离心操作结束后, 室温分离血浆。分离的血浆样品冷冻保存于超低温冰箱(冰箱温度范围不高于-60 ℃)中,用于检测血浆中阿普斯特的血药浓度。
2 方法与结果
2.1 方法学验证[11]
2.1.1 线性范围用移液器取285 μL空白血浆,加入15 μL不同浓度的分析物工作溶液,使样品中阿普斯特的浓度分别为2.00、4.00、10.0、30.0、100、200、480、600 ng·mL-1。分别按“1.4”项下方法处理样品,按“1.2”项下条件测定。每个分析批的标准曲线采用双校正样品拟合,以阿普斯特与阿普斯特-d5的色谱峰面积比为纵坐标(Y),以血浆中阿普斯特的浓度为横坐标(X,μg·mL-1),采取加权(W=1/X2)最小二乘法进行线性回归,求得回归方程为Y=0.014 798 2X-0.004 368 50,决定系数r2为0.997 7,校正样品回算浓度均在要求范围之内,说明阿普斯特在2.00~600 ng·mL-1范围内线性良好,由此计算的药物浓度准确可靠。本研究方法的定量下限为0.200 μg·mL-1,信噪比S/N>10,满足定量要求。
2.1.2 残留通过在注射高浓度样品后注射空白基质样品(空白血浆)来评价残留,图A为阿普斯特在定量上限(600 ng·mL-1)处的选择离子色谱图,分析物和内标经色谱分离后,分析物(阿普斯特)的保留时间为 1.2 min,内标(阿普斯特-d5)的保留时间为 1.2 min,并得到较好的响应值与峰形。图 B为注射定量上限(600 ng·mL-1)阿普斯特样品后进样的空白血浆样品色谱图。将图A与图B进行比较,发现空白血浆样品的色谱图在阿普斯特与阿普斯特-d5的色谱峰附近中没有干扰峰出现,说明该方法的残留小,对分析物(阿普斯特)及内标(阿普斯特-d5)的定量均无影响。图C为受试对象服用阿普斯特片4 h后的血浆样品提取离子色谱图,结果表明该研究方法在用于测定实际血浆样品时相应保留时间处无飘移。
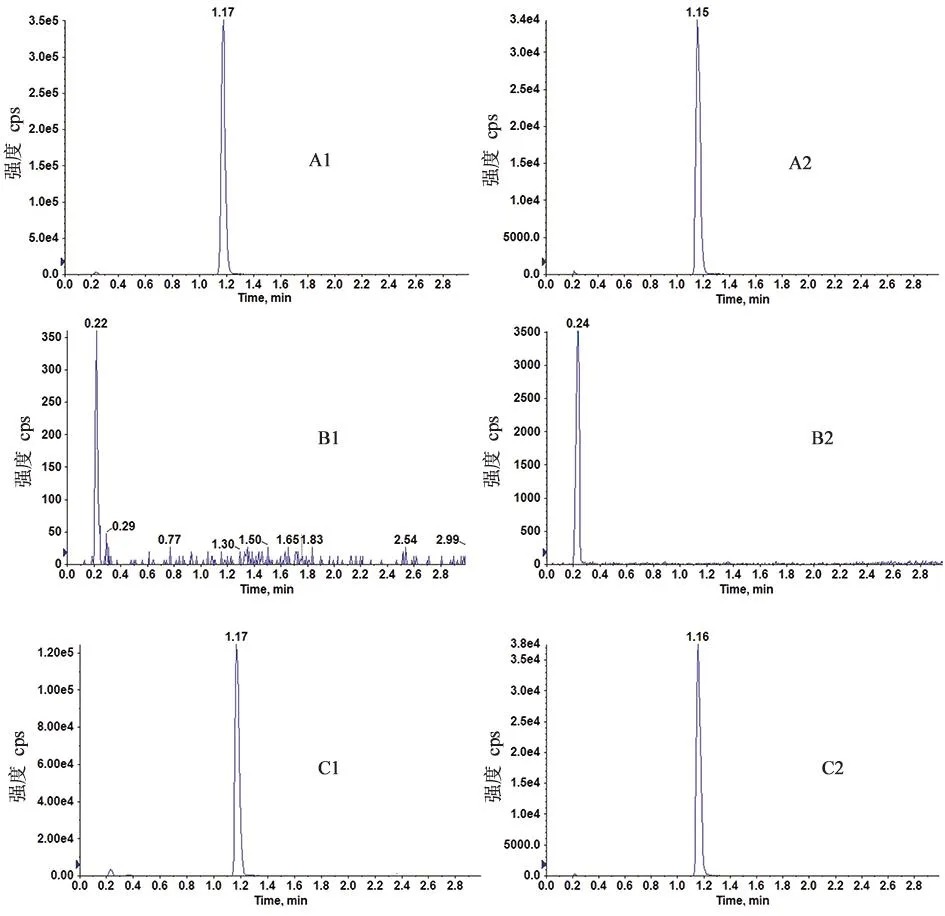
A.加标阿普斯特的血浆样品;B.空白血浆样品;C.受试者服用阿普斯特片4 h后的血浆样品 1.阿普斯特;2.内标
2.1.3 精密度与准确度配制含分析物(阿普斯特)定量下限(2.00 ng·mL-1)和低(6.00 ng·mL-1)、中(50.0 ng·mL-1)、高(450 ng·mL-1)水平的质控样品各 6份,“1.4”项下方法处理样品,在至少2 d内测定3个不同分析批,评价本研究方法的批内、批间准确度和精密度;结果见表 4。表明该研究方法的精密度、准确度均良好,符合《中国药典》2020年版(四部)《通则9012生物样品定量分析方法验证指导原则》。
2.1.4 选择性取6个受试者的空白血浆制备空白基质样品与定量下限样品,空白基质样品除用300 μL 乙腈溶液代替内标工作溶液外,其余步骤按“1.4”项下方法处理样品,定量下限样品按“1.4”项下方法处理样品,采用“1.2”项下条件进样分析。本研究6个受试者的空白基质干扰组分在阿普斯特与阿普斯特-d5保留时间处的响应均为0,表明该方法具有良好的选择性,能区分阿普斯特与阿普斯特-d5与基质中的内源性组分。

表4 准确度与精密度(n=6)
2.1.5 基质效应取6个受试者的空白基质、1个高血脂空白基质及1个溶血空白基质制备不添加待测物和内标的空白基质样品,“1.4”项下方法处理样品,向处理后的空白基质样品中分别加入低质控浓度(LQC,6.00 ng·mL-1)和高质控浓度(HQC,450 ng·mL-1)阿普斯特与阿普斯特-d5的混合溶液,配制基质效应评价样品,采用“1.2”项下条件进样分析,所得阿普斯特与阿普斯特-d5响应与相同理论浓度的纯溶液样品的阿普斯特与阿普斯特-d5响应比较,用内标归一化的基质因子的变异系数(CV)评价基质效应,具体结果见表5。结果6个受试者,1个高脂及1个溶血的空白基质样品的内标归一化基质因子的CV均小于15%,表明该研究方法的基质效应符合要求。
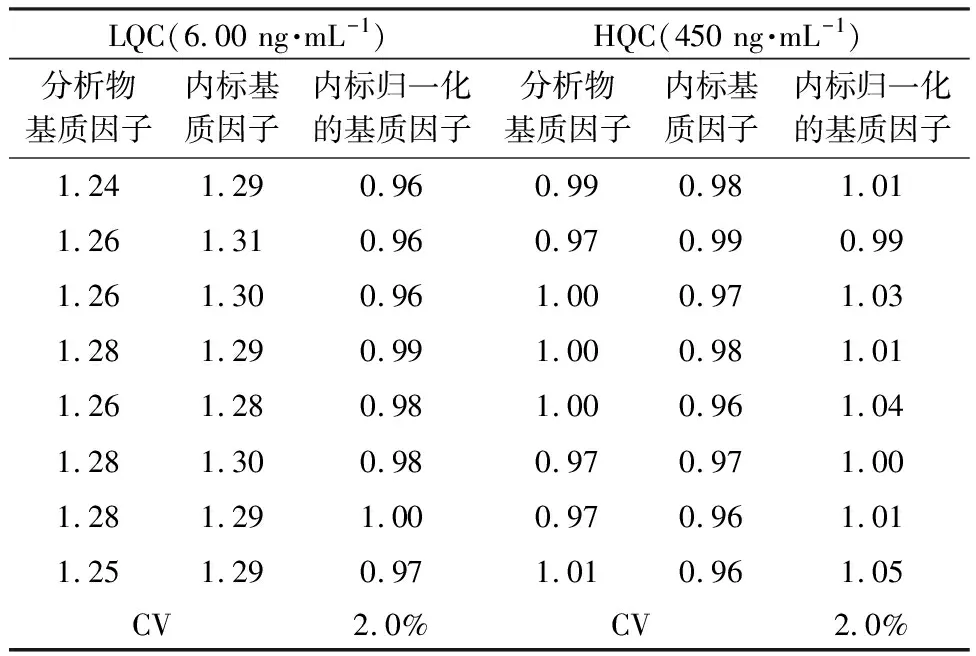
表5 6批不同来源血浆、高脂及溶血的基质效应
2.1.6 提取回收率取空白基质,按“1.4”项下方法处理样品,加入阿普斯特与阿普斯特-d5,进样测定,比较二者的响应均值,考察阿普斯特(低、中、高 3个质控浓度)和阿普斯特-d5的提取回收率。3种质控浓度阿普斯特的提取回收率分别为94.3%、93.2%、 94.0%,阿普斯特-d5的提取回收率为96.7%,说明该研究方法提取回收率较高,在处理过程中不会对结果造成影响。
2.1.7 稳定性用新鲜制备的标准曲线及质控样品,考察放置不同储存条件下的样品中阿普斯特的稳定性,分别考察了含阿普斯特的血浆样品反复冻融循环5次的稳定性、血浆样品在室温条件下放置25 h的稳定性、处理过的血浆样品在自动进样器温度(8 ℃)条件下放置96 h的稳定性及血浆样品在冷冻条件下(-80 ℃,-20 ℃)放置62 d的稳定性。结果见表6。结果表明阿普斯特血浆样品在考察条件下稳定。
表6 不同条件下的稳定性考察结果
2.2 阿普斯特片药代动力学及生物等效性研究使用本研究建立的UPLC-MS/MS方法对血浆中阿普斯特浓度进行测定,评价空腹和餐后条件下两制剂之间是否具有生物等效性。以时间为横坐标、测定的阿普斯特血药浓度为纵坐标,分别绘制空腹及餐后条件下受试制剂与参比制剂的阿普斯特平均血药浓度-时间曲线(见图2)。

图2 在空腹(A,n=12)和餐后(B,n=12) 条件下阿普斯特的平均血药浓度-时间曲线图
用Phoenix WinNonlin 8.2软件计算两种阿普斯特片剂的药代动力学参数(AUC0~t、 AUC0~∞、Cmax、λz、 t1/2、Tmax),结果见表7。主要药代动力学参数(AUC0~t、 AUC0~∞、Cmax)经对数转换后进行多因素方程分析,同时采用(1-2ɑ)计算两种制剂的主要药代动力学参数的几何均值的90%置信区间(CI),评价两种制剂的生物等效性。12例受试者空腹口服阿普斯特片受试制剂和参比制剂的Cmax、AUC0~t、AUC0~∞几何均值比的90%置信区间分别落在92.52%~112.38%、96.25%~110.28%和96.15%~110.52%范围内,30例受试者餐后口服阿普斯特片受试制剂和参比制剂的Cmax、AUC0~t、AUC0~∞几何均值比的90%置信区间分别落在93.56%~114.85%、96.57%~116.20%和99.73%~114.38%范围内。此外,本实验中空腹给药后阿普斯特Cmax、AUC0~t、AUC0~∞的个体内变异CV分别为13.25%、9.55%、9.48% ;餐后给药组的个体内变异CV分别为15.66%、14.21%、14.62%。AUC0~t、 AUC0~∞和Cmax经对数转换后多因素方差分析结果显示给药周期、给药序列、个体间、制剂间的差异均具有统计学意义(P<0.05)。
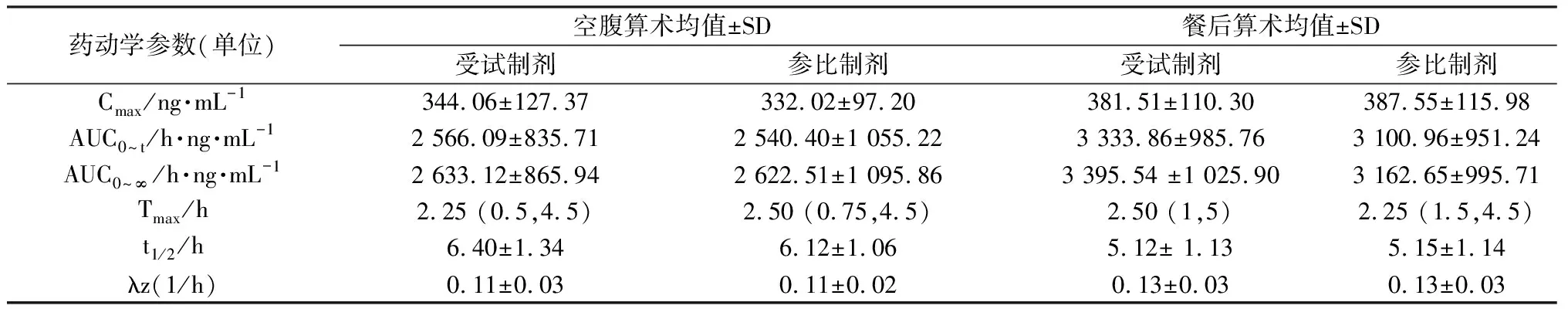
表7 在空腹和餐后条件下阿普斯特片受试制剂和参比制剂药动学参数(n=12)
3 结论
本研究建立的测定人血浆中阿普斯特含量的方法具有灵敏度高、准确可靠、选择性好、基质效应小、快速简便等优点。该方法使用乙腈对血浆样品进行蛋白沉淀,内标工作溶液直接配制到沉淀剂中,减少了操作步骤,使操作更简化,并且有机试剂消耗少,大大提高了临床适用性和工作效率;选用0.1%甲酸水和0.1%甲酸乙腈为流动相,提高灵敏度,出峰位置稳定,且缩短了单针检测时间;该方法使用同位素标记的内标,方法的耐用性大大提高;此外为了减小该分析方法的基质效应,将蛋白沉淀离心后的上清进行1∶1稀释(稀释液:10%乙腈水)。经方法学验证该方法符合生物样本分析要求,实际样本再分析的通过率为100%,将该方法成功应用于临床血药浓度检测,结果可靠。本试验为阿普斯特相关制剂的生物等效性研究提供了数据参考,适用于阿普斯特的血药浓度检测及其药代动力学研究,为阿普斯特制剂一致性评价提供依据。
研究结果表明,空腹和餐后口服阿普斯特片受试制剂和参比制剂的Cmax、AUC0~t、AUC0~∞几何均值比的90%CI均在80.00%~125.00%等效区间内(包括边界值),符合生物等效性评价标准。此外,本试验中空腹和餐后给药后阿普斯特Cmax、AUC0~t、AUC0~∞的个体内变异CV均小于30%,不属于高变异药物。Cmax、AUC0~t、AUC0~∞经对数转换后的多因素方差分析结果显示给药周期、给药序列、个体间、制剂间的差异均具有统计学意义(P<0.05)。研究结果表明两种制剂在空腹和餐后条件下口服给药符合生物等效。另外,由药动学参数AUC0-t可知阿普斯特餐后条件下口服给药生物利用度较空腹条件下高,为临床提供更合理的用药指导,为患者提供更好的治疗效果。
