我国化学药生产企业境外检查观察情况分析
2024-02-25徐长波翟铁伟
徐长波,翟铁伟
(国家药品监督管理局食品药品审核查验中心,北京 100044)
随着我国科学技术的迅猛发展,以及市场发展及政府配套措施的扶助引导,医药工业转型升级[1],我国已形成较完善的制药产业链和产业群[2]。众多医药企业正在走向国际化经营模式[3-4],并具备了一定的全球竞争力,越来越多的境外药品监管机构也来华进行药品检查。20世纪90年代末,我国开始了对来华进行药品检查的境外药品监管机构的相应检查观察工作。2010 年前后,原国家食品药品监督管理局药品认证管理中心逐渐将境外检查观察工作制度化,我国药品监管部门也开始加速推进药品检查国际化,不断提升药品检查能力。随着境外药品监管机构来华检查的日益增加,国家药品监督管理局(NMPA)于2019 年进一步完善了境外检查观察的相应管理。本研究中通过分析2015 年至2022 年我国化学药生产企业境外检查观察情况及检查发现缺陷分布,总结境外检查重点和高频缺陷的类别。现报道如下。
1 药品境外检查观察
当境外药品监管机构对我国人用药品进行检查时,我国药品监督管理部门会派出具有经验的药品检查员对境外药品监管机构的检查过程进行见证和记录。根据NMPA与境外药品监管机构签署的相关合作协议,境外药品监管机构派员对我国企业开展检查时,通常会通知NMPA 派出观察员进行观摩。NMPA 食品药品审核查验中心(简称核查中心)具体负责境外检查观察的具体实施。当核查中心接到NMPA境外药品监管机构检查计划或通知后,会选派有资质的药品检查员作为境外检查的观察员。观察员对检查的全过程进行观察并客观记录,包括文件记录、现场情况、提出的问题和建议等,并书写观察检查报告。
2 2015 年至2022 年我国化学药接受境外检查观察概况
2.1 总体情况
2015年至2022年,美国食品和药物管理局(FDA)、世界卫生组织(WHO)、欧洲药品质量理事会(EDQM)、德国汉堡健康及消费者保护部(BGV)、巴西卫生监督局(ANVISA)等24 个境外药品监管机构检查我国化学药生产企业443家次。
2.2 检查次数
2015 年至2019 年,境外药品监管机构检查数量逐年增加,分别为74,81,84,82,102 家次;受新型冠状病毒感染(简称新冠)疫情影响,2020年至2022年,境外检查数量急剧减少,分别为6,6,8 家次,其中远程检查分别为3,3,4家次。详见图1。
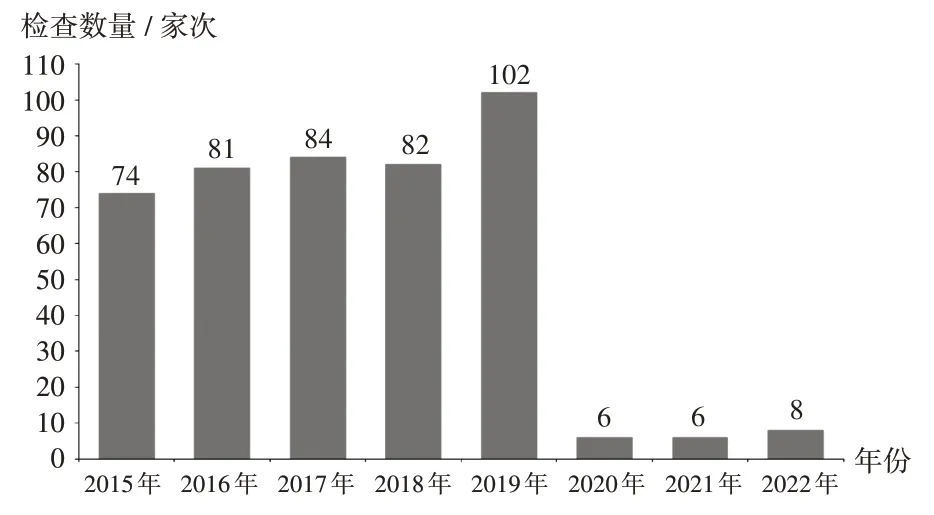
图1 2015 年至2022 年境外检查观察次数Fig.1 Number of international drug inspection and observation from 2015 to 2022
2.3 检查发现的缺陷
随着《药品生产质量管理规范》(2010 年修订,以下简称GMP)的深入实施,我国药品生产企业整体GMP水平不断提高[5]。2015年至2022年,核查中心共进行443家次境外检查观察,记录缺陷4 854 项;境外药品监管机构检查发现的次均缺陷数呈下降趋势。详见图2。
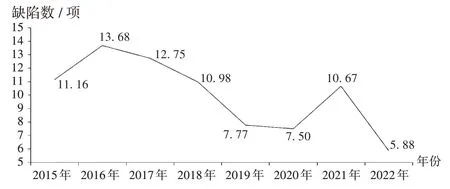
图2 2015 年至2022 年我国化学药生产企业境外检查观察记录缺陷均值Fig.2 Average values of defects recorded by international drug inspection and observation of Chinese chemical pharmaceutical production enterprises from 2015 to 2022
由于不同境外药品监管机构的检查员背景和专长有差异,所发现缺陷数量存在差异。2015 年至2019 年,WHO 和EDQM 检查发现的缺陷数量相对较多,平均每次检查发现约20 项,且所有问题均进行详细描述;FDA检查发现的缺陷数量相对较少,平均每次检查发现约5 项,由检查员结合产品风险和发现缺陷进行综合判断后形成最终缺陷项。详见图3。
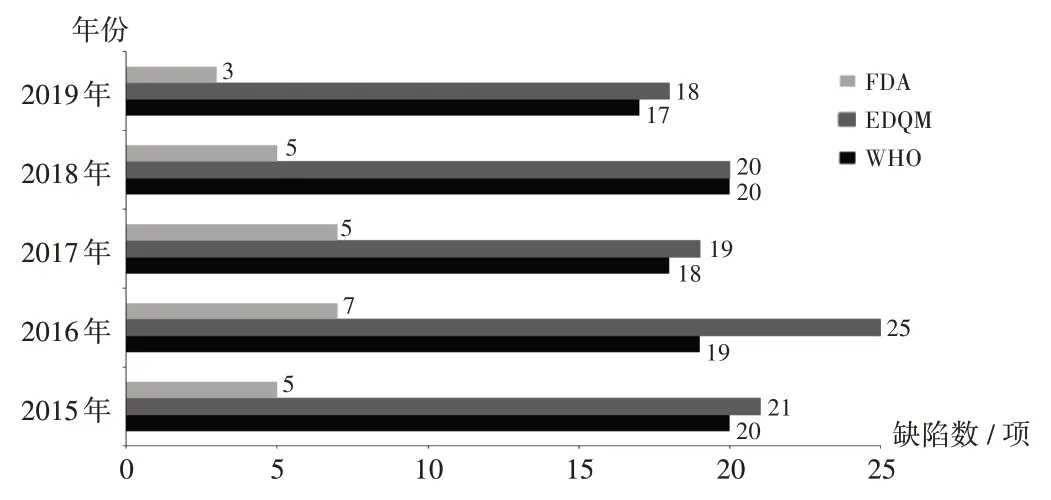
图3 2015 年至2019 年WHO,EDQM,FDA 检查观察记录的平均缺陷Fig.3 Average number of defects recorded by WHO,EDQM,FDA inspection observations from 2015 to 2019
受新冠疫情影响,2020 年至2022 年的境外检查数量明显减少。俄罗斯联邦药物与规范研究院(SID&GP)、波兰药品检查局(CPI)采用远程检查的方式进行检查,FDA 和WHO 等仍进行现场检查。2020 年至2022 年,现场检查10次,发现缺陷87项;远程检查10次,发现缺陷44项。
2.4 检查发现的严重缺陷
2015 年至2019 年,境外检查发现严重缺陷的次数占比逐年降低,发现的严重缺陷主要由数据可靠性问题引起;2021年至2022年,均未发现严重缺陷。详见表1。

表1 2015 年至2019 年境外检查观察记录我国化学药生产企业的严重缺陷Tab.1 Critical defects found during drug international drug inspection and observation from 2015 to 2019
数据可靠性问题可简单分为规范性因素和真实性因素两方面[6]。2015年至2017年,严重缺陷主要是违背诚实信用原则故意编造、篡改数据等导致的数据可靠性问题,随着全球主要药品监管机构、国际组织和我国药品监管部门针对药品数据发布的一系列指南、法规,我国药品生产企业质量管理体系不断提高,真实性因素导致的数据可靠性缺陷逐渐被规范性因素导致的数据可靠性替代[7]。2018年至2022年,境外检查发现的严重缺陷开始转向了无菌保障、风险评估、变更控制、防交叉污染等多个影响产品质量的关键因素。
3 2019 年化学药生产企业接受境外检查观察分析
3.1 我国核查中心检查观察情况
3.1.1 检查观察次数
2019 年,核查中心组织开展对主要包括FDA 等在内的13 个境外药品检查机构的检查观察102 家次,其中FDA,WHO,EDQM 3 个境外药品检查机构检查观察82家次,约占全年检查观察的80.39%。详见图4。
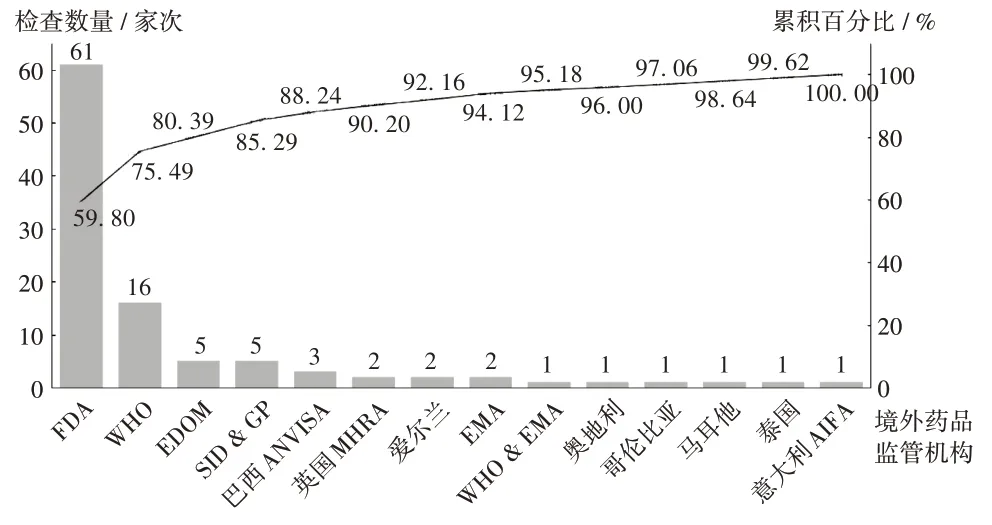
图4 2019 年境外检查观察情况Fig.4 Number of international drug inspection and observation in 2019
3.1.2 检查观察发现的缺陷
2019 年,检查观察共记录境外药品检查机构检查发现缺陷793 项,参照GMP 正文章节对境外检查观察中记录的缺陷进行分析。其中,质量控制与质量保证、文件管理、确认与验证、设备、生产管理5个类别的缺陷约占全部缺陷的77.43%。详见图5。

图5 2019 年境外检查观察记录我国化学药生产企业的缺陷分布Fig.5 Distribution of defects found during international drug inspection and observation of Chinese chemical pharmaceutical production enterprises in 2019
3.2 境外药品监管机构检查观察情况
3.2.1 FDA
2019 年,核查中心共组织FDA 对我国化学药生产企业检查观察61家次,发现缺陷197项,约占2019年全部境外药品监管机构发现缺陷的24.84%,平均每次检查提出缺陷约3项。参照GMP正文章节,将FDA 检查观察中记录的缺陷分类,详见图6 A。质量控制与质量保证、文件管理、确认与验证部分提出的缺陷占比分别居第1,2,3 位,占比分别为40.61%,18.78%,10.15%,与2015年至2018年FDA在我国检查发现的情况一致[8]。
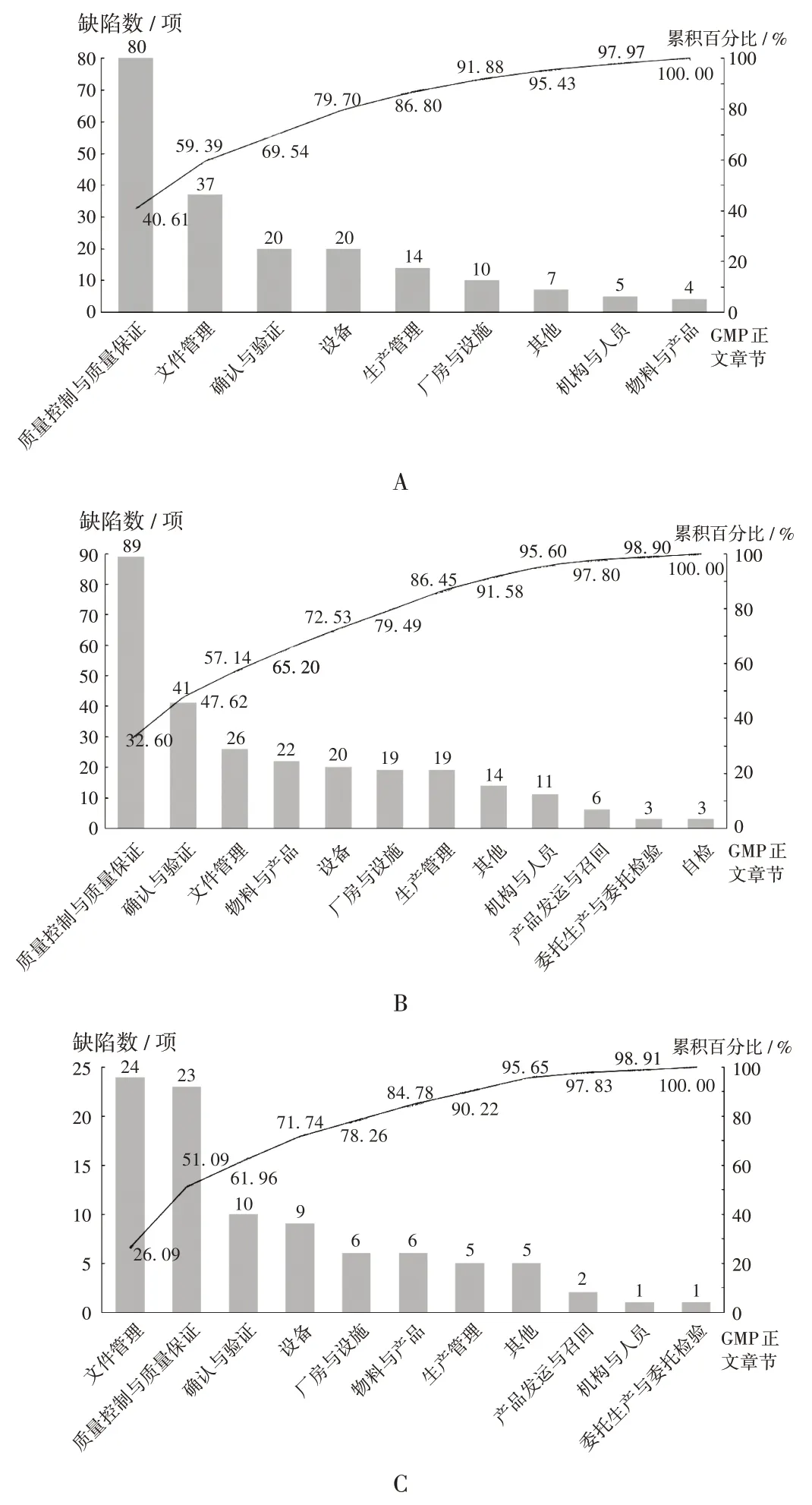
A.FDA B.WHO C.EDQM图6 2019 年部分境外监管机构检查观察记录我国化学药生产企业的缺陷分布A.FDA B.WHO C.EDQMFig.6 Distribution of drug inspection and observation defects of Chinese chemical pharmaceutical production enterprises by some overseas pharmaceutical regulatory agencies in 2019
1)质量控制与质量保证问题涉及偏差调查不充分/未找到根本原因,纠正措施和预防措施(CAPA)无效,变更的风险评估不完整,年度质量回顾不全面,投诉未启动调查,稳定性考察项目的方法/标准与《美国药典》(USP)不一致,仪器缺少/未开启审计追踪,检测仪器多人共用一个账户,取样不具代表性,检验的原始记录/附图不完全,质量管理部门对电子数据审核缺失等。
2)文件管理问题涉及记录的信息不完整/缺失,记录的唯一性不受控,记录未及时填写和签名,关键的操作/数据等缺少签名和复核,相关操作未建立规程等。
3)确认与验证问题涉及清洁验证不足,部分设备/器具的清洁程序未验证,工艺验证未涵盖工艺参数范围,设备确认未涵盖生产使用范围等。
3.2.2 WHO
2019 年,核查中心共组织WHO 对我国化学药生产企业检查观察16家次,发现缺陷273项,约占2019年全部境外药品检查机构发现缺陷的34.43%,平均每次检查提出缺陷约17 项。参照GMP 正文章节,对WHO 检查观察中记录的缺陷分类,详见图6 B。质量控制与质量保证、确认与验证、文件管理部分提出的缺陷分别居第1,2,3位,占比分别为32.60%,15.02%,9.52%。
1)质量控制与质量保证问题涉及偏差未按流程调查及CAPA 不足,偏差未定期回顾,缺少审计追踪功能或未启用,时区未锁定/ 各检测仪器时间存在较大差异,分析方法变更后未验证,水、环境检测、投诉、偏差等未进行年度回顾,变更未进行风险评估/未对变更前后进行对比,微生物实验室的标识、人员培训、灭菌设备等管理存在不足,未进行培养基适应性试验等。
2)确认与验证问题涉及验证未考虑最差条件,未建立(再)验证与确认计划,验证与确认未进行/部分遗漏,未进行清洁验证/清洁验证存在清洁溶剂选择错误,未考虑残留及擦拭点未评估,工艺验证不充分,溶剂回收风险评估不足,物料/中间体等制订的效期未验证等。
3)文件管理问题涉及批记录设计不全,批记录未记录偏差等异常事件,不同文件/记录不一致,规程与实际操作不一致,记录散页不受控,未及时记录和复核等。
3.2.3 EDQM
2019 年,核查中心共组织EDQM 对我国化学药生产企业检查观察5家次,发现缺陷92项,约占2019年全部境外药品检查机构发现缺陷的11.60%,平均每次检查提出陷约18 项。参照GMP 正文章节,对EDQM 检查观察中记录的缺陷分类,详见图6 C。文件管理、质量控制与质量保证、确认与验证部分提出的缺陷分别居第1,2,3位,占比分别为26.09%,25.00%,10.87%。
1)文件管理问题涉及批记录未记录偏差等异常事件,文件规定不全/未建立相关规程,文件规定不合理/错误,记录设计不全/信息填写不全,活页记录不受控,记录编号/页码控制缺失,操作人员/复核人员未签名等。
2)质量控制与质量保证问题涉及偏差调查不彻底、评估不充分,变更未进行评估/变更未完成前已实施,年度质量回顾不全,水系统、洁净区环境等监测缺乏审查和趋势分析,新增供应商未确认,取样点未评估/检测的样品不具代表性,计算机未得到适当控制等。
3)确认与验证问题涉及工艺验证考虑不全,计算机系统验证未考虑操作系统的更新问题,执行的参数、效期等没有进行验证,设备验证不包括附属设施,灭菌验证温度探头放置位置不科学等。
4 建议
通过分析近年来我国化药品生产企业境外检查观察情况及缺陷分布,发现不同境外药品检查机构虽发现的具体缺陷有差异,但检查的重点及发现高频缺陷的类别基本类似,表明我国化学药生产企业在质量控制和质量保证、确认和验证、文件管理等方面仍与国际水平有一定差距,建议药品生产企业对症下药,全面提高质量管理水平。另外,基于化学药生产企业GMP 的实际情况,我国药品检查机构也应将质量控制和质量保证、确认和验证、文件管理等作为检查重点,结合化学药生产企业的具体情况,有的放矢地进行检查。
质量控制和质量保证、确认与验证、文件管理等是药品检查发现缺陷的“重灾区”。现阶段,我国相当部分的化学药生产企业以符合法规为目的、按GMP 组织生产活动,但未将GMP 作为强化自身管理的有力工具。化学药生产企业应根据自身实际,活学活用,持续提升其生产质量管理水平,最终形成企业独特的质量文化[9-10]。
我国药品生产企业GMP 水平参差不齐,而检查资源又相对有限[11-12]。我国药品监管机构可在境外药品检查机构监管的基础上进行风险研判,聚焦检查关键性问题及高风险问题。同时,对境外药品检查机构监管发现的问题深入总结与分析,作为了解我国化学药品行业生产质量管理水平的重要手段。同时,与我国药品检查机构进行药品监管时发现的问题进行对比分析,深入了解药品的行业现状和检查现状,做到基于风险的药品检查[13-14]。
