国产艾司唑仑原料药及其片剂关键质量属性研究进展*
2024-02-25沈丹丹胡振晶曾令高
沈丹丹,胡振晶,曾令高,唐 华△
(1.重庆市食品药品检验检测研究院·国家药品监督管理局麻醉精神药品质量监测重点实验室,重庆 401121;2.重庆大学生物工程学院,重庆 400044)
1 产品关键质量指标
1.1 国产艾司唑仑原料药合成工艺
国产艾司唑仑原料药合成工艺主要包括2条路线,均以2-氨基-5-氯-二苯甲酮为起始物料,经过一系列反应制得艾司唑仑粗品,再经精制结晶得到成品,2 条合成路线主要差异为艾司唑仑母环结构中三氮唑环与苯并二氮杂环的环合顺序不同。由2 条合成路线可知,艾司唑仑原料药引入的起始物料、中间体及反应副产物较多,且涉及的部分反应试剂为基因毒性杂质,故杂质谱是影响本品安全性的关键质量属性指标。
艾司唑仑粗品合成工艺路线1:以2-氨基-5-氯-二苯甲酮为起始物料,经甲脒反应制得甲脒物(3-氨基物),再经肼化反应生成酰肼,酰肼通过酰化反应得艾司唑仑酰化物(氯乙酰氨基氯二苯酮),再经重排得二苯酮,成盐后与乌洛托品环合制得艾司唑仑粗品。详见图1 A。有2家生产企业分别以中间体甲脒物(3-氨基物)和艾司唑仑酰化物(氯乙酰氨基氯二苯酮)作为起始物料,再经路线1的酰化反应、环合反应制得艾司唑仑粗品。
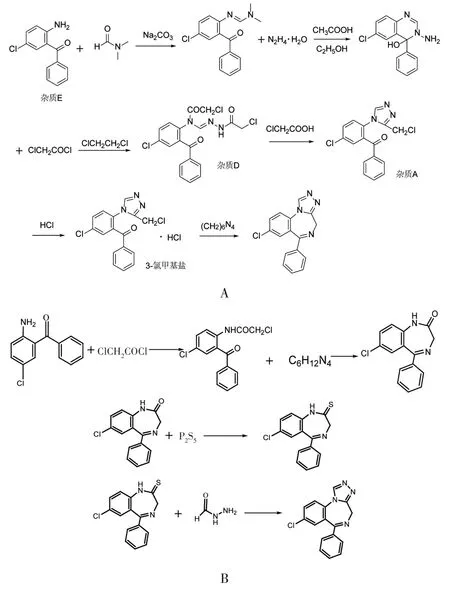
A.路线1 B.路线2图1 艾司唑仑粗品合成工艺A.Route 1 B.Route 2Fig.1 Synthesis process of estazolam
艾司唑仑粗品合成工艺路线2:以2-氨基-5-氯-二苯甲酮为起始物料,经酰化反应制得艾司唑仑酰化物(氯乙酰氨基氯二苯酮),通过与乌洛托品环合反应生成艾司唑仑环合物(去甲西泮),再与五硫化二磷硫化反应生成艾司唑仑硫化物,最后通过与甲酰肼反应制得艾司唑仑粗品。详见图1 B。
1.2 国产艾司唑仑片处方工艺
目前,上市销售的艾司唑仑片均为素片,均采用湿法制粒工艺制备。通过一致性评价的4家生产企业的处方组成与参比制剂基本一致,原料药均控制了粒度,但限度要求不同,辅料主要为玉米淀粉(填充剂)、羟丙纤维素(崩解剂)、硬脂酸镁(润滑剂)、乳糖水合物(填充剂),仅1 家生产企业处方中黏合剂为低取代羟丙纤维素;未通过一致性评价的生产企业处方组成差异较大,部分生产企业对原料药进行了粉碎,但均无粒度控制要求,辅料中填充剂、黏合剂、崩解剂种类和用量不同,有4家生产企业使用乙醇作为润湿剂。本品生产工艺较简单,规格小,辅料占比高,工艺过程控制可能对产品含量均匀度有显著影响。可见,含量均匀度、溶出度及含量测定是影响本品有效性及均一性的关键质量属性指标。
1.3 产品质量风险评估
根据原料药合成工艺及制剂处方工艺,结合艾司唑仑原料药及其片剂最新国内外质量标准收载情况,对质量指标进行风险分析,确定影响产品安全性、有效性及均一性的关键质量属性指标。详见表1。

表1 艾司唑仑原料药和片剂关键质量属性指标的风险评估Tab.1 Risk assessment of critical quality attribute indicators of estazolam API and Estazolam Tablets
2 关键质量属性指标控制方法
2.1 安全性
2.1.1 有关物质
根据国内艾司唑仑原料药合成工艺,粗品合成路线最长的为6步反应,最短的为2步反应。共涉及3个起始物料,包括2-氨基-5-氯-二苯甲酮、甲脒物(3-氨基物)和艾司唑仑酰化物(氯乙酰氨基氯二苯酮),其中甲脒物和艾司唑仑酰化物又为中间体,根据起始物料、中间体、反应副产物、降解产物等共推导出11 个杂质。详见表2。

表2 根据原料药合成工艺推导的艾司唑仑杂质谱Tab.2 Impurity spectrum of estazolam derived from the synthesis process of estazolam API
除JP18及《日本药典外药品标准(第三部)》有关物质采用薄层色谱法外,其余质量标准均采用高效液相色谱(HPLC)法。艾司唑仑原料药有关物质控制情况:1)ChP2020(二部)艾司唑仑原料药及其片剂有关物质采用不加校正因子的自身对照法计算,色谱条件一致,以十八烷基硅烷键合硅胶为填充剂,流动相为甲醇-水(65∶35,V/V),检测波长为223 nm,供试品溶液质量浓度为0.2 mg/mL,原料控制总杂质限度为0.5%,片剂控制总杂质限度为1.0%,缺失已知杂质控制。2)USP2022 原料药有关物质采用加校正因子的主成分外标法计算,以十八烷基硅烷键合硅胶为填充剂,流动相为乙腈-水(梯度洗脱),检测波长为254 nm,供试品溶液质量浓度为1 mg/mL,控制了5个已知杂质,限度均为0.1%,未知单个杂质的限度为0.10%,总杂质限度为0.5%。
艾司唑仑片有关物质控制情况:1)USP2022艾司唑仑片有关物质采用峰面积归一化法计算,以苯基硅烷键合硅胶为填充剂(150 mm×4.6 cm,3µm),以质量浓度为2.8 g/L 的磷酸二氢钾溶液(用1 mol/L 氢氧化钠溶液调pH至6.5)-甲醇-乙腈(55∶35∶10,V/V/V)为流动相,检测波长为254 nm,供试品溶液质量浓度为0.02 mg/mL,单个杂质和总杂质限度分别为0.5%和1.0%。2)艾司唑仑片一致性评价后的质量标准如下,YBH03582022有关物质色谱条件与ChP2020基本一致,仅检测波长为254 nm;YBH06222021 和YBH08682021与USP2022 艾司唑仑片色谱条件一致;YBH09462021质量标准流动相与USP2022 艾司唑仑片一致,但采用梯度洗脱,杂质控制最严格,5 个已知杂质、单个杂质、总杂质限度分别为0.2%,0.2%,1.0%。可见,各标准有关物质色谱条件、供试品溶液质量浓度的差异可能导致杂质检测能力和分离能力不同。
范钢[19]对艾司唑仑中间体合成工艺及有关物质结构确证进行研究,对1 个艾司唑仑中间体及6 个有关物质的结构进行解析,但并未对这7个杂质的控制方法进行研究。且目前未见其他文献报道艾司唑仑引入的10个相关杂质的全面控制方法,通过对比各标准色谱条件,发现质量标准YBH09462021 能有效分离8 种杂质,而其他标准均存在杂质共同洗脱的问题。
2.1.2 基因毒性杂质
根据艾司唑仑原料药合成工艺,2-氨基-5-氯-二苯甲酮的起始物料为对氯硝基苯,环合反应使用的试剂乌洛托品的起始物料为甲醛,还使用了甲酰肼、氯乙酰氯等含有警示结构的基因毒性杂质作为反应试剂,通过全面考虑起始物料、试剂、溶剂和副产物等可能引入的基因毒性杂质,累积评估8 个基因毒性杂质(表3)。对于无毒理学数据的基因毒性杂质,按允许暴露量(PDE)为1.5 µg/d,艾司唑仑每日最大服用剂量为12 mg,限度计算为125×10-6。本品基因毒性杂质种类多、结构差异大,且多数杂质缺乏检测响应结构,对分析仪器的灵敏度、准确度和选择性要求较高。虽然目前国内外未见相关文献报道艾司唑仑原料药中基因毒性杂质含量的研究,但有较多文献报道了药物中甲酰肼、氯乙酰氯、氯乙酸、N-亚硝胺类等基因毒性杂质的研究[23-31]。故以此建立了测定艾司唑仑原料药中甲醛含量的HPLC衍生法,发现1家生产企业的2批原料药中甲醛含量达60×10-6,远超过控制阈值的33×10-6(30%限度浓度),建议生产企业将甲醛纳入质量标准,并依据多批次原料积累数据,制订合理的控制策略。
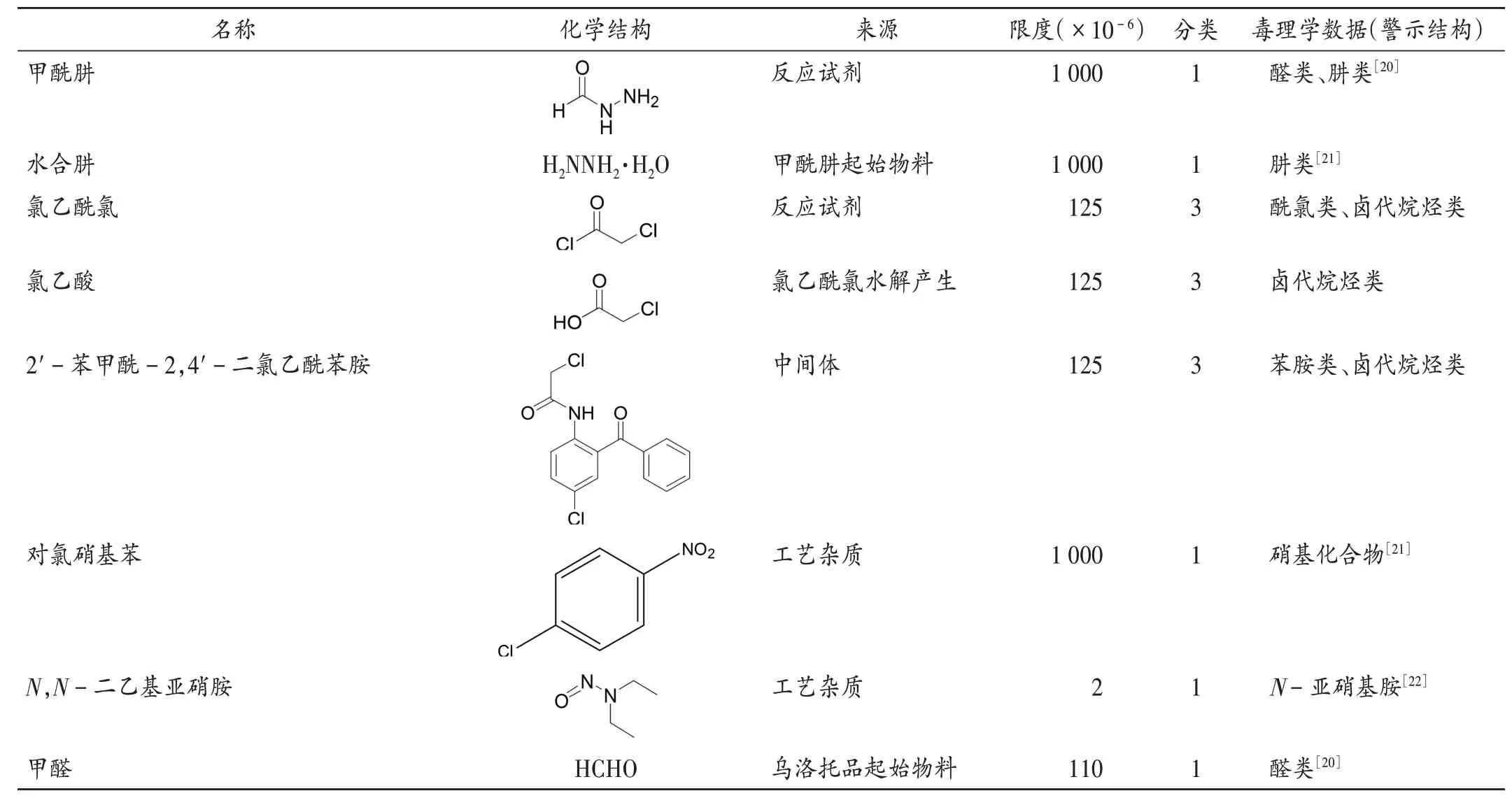
表3 艾司唑仑原料药基因毒性杂质Tab.3 Genotoxic impurities of estazolam API
2.1.3 残留溶剂
艾司唑仑原料药合成工艺中使用了12 种有机溶剂,包括环己烷、乙醇、乙腈、正丁醇、二氯甲烷、乙酸乙酯、甲醇、三乙胺、二氯乙烷、二甲基甲酰胺、冰醋酸、苯。其中,1 类溶剂5 种,2 类溶剂2 种,3 类溶剂5 种。同时,艾司唑仑片未通过一致性评价的4家生产企业制粒工艺均使用乙醇作为润湿剂。前期调研发现,4 家生产企业均未考察乙醇残留量。ChP2020艾司唑仑原料药中未规定需要检测的残留溶剂种类及方法,4 个原料药生产企业所用残留溶剂不同,限定残留溶剂种类的方法可能不具有通用性。USP2022 与《欧洲药典11.2 版》(EP11.2)对残留溶剂的控制方法相似,通则中提供的方法对残留溶剂采用分类控制的策略,将供试品中的残留溶剂按1 类、2 类、3 类残留溶剂分别进行控制。参考ChP2020(四部)通则0861 残留溶剂项下方法,分别采用极性色谱柱HP-INNOWAX 柱(30 m × 0.32 µm,0.5 µm)和非极性色谱柱DB-1 柱(30 m × 250 µm,0.5µm),以丁酮为参比溶剂,用程序升温方法,测定艾司唑仑原料药及其片剂中残留溶剂的保留时间。结果2 家生产企业的艾司唑仑片中检出与乙醇校正相对保留时间(RART)一致的色谱峰,并建立乙醇定量测定方法,结果8批样品中的乙醇含量为0.4%~0.5%。
2.2 有效性
2.2.1 溶出度
艾司唑仑属基于药物体内分布的生物药剂学分类系统(BDDCS)2 类药物,即低溶解性强代谢性药物。伏圣青[32]采用Caco-2 体外细胞模型对艾司唑仑的渗透性进行了研究,认为艾司唑仑属生物药剂学分类系统(BCS)1类药物,即高溶解性高渗透性药物,推测主要是考虑到艾司唑仑的剂量规格,故BDDCS 与BCS 分类不同。虽然各标准溶出度限度均为标示量的80%,但ChP2020 溶出度条件与其他标准差异显著。ChP2020中,艾司唑仑片溶出条件采用第三法(小杯法),转速为100 r/min,溶出介质为盐酸溶液(9→1 000)100 mL(1 mg 规格)或200 mL(2 mg 规格),取样时间为30 min,样品在此条件下溶出较剧烈,不能有效评价制剂的溶出情况;采用紫外光谱(UV)吸收系数法测定溶出量,存在专属性不高的问题。一致性评价后,质量标准中溶出度与USP2022及《日本药典外药品标准(第三部)》一致,均采用桨法,溶出介质为水900 mL,转速为50 r/min,时间为30 min,采用HPLC 对照品外标法测定溶出量。伏圣青等[33]采用桨法进行溶出度试验,并建立了灵敏度更高的HPLC 外标法,可反映不同生产企业不同批次间艾司唑仑片溶出行为的差异,发现国内艾司唑仑片与参比制剂在溶出介质水中溶出行为差异较大。参照日本橙皮书收载的艾司唑仑片溶出曲线条件,发现通过一致性评价的4 家生产企业的艾司唑仑片的溶出行为与参比制剂一致,在4 种溶出介质中,15 min 时累积溶出量达85%,为快速溶出制剂。通过一致性评价的7家生产企业的艾司唑仑片15 min 时累积溶出量未达到85%,且与参比制剂溶出曲线计算的相对校正因子(f2)值均小于50,未满足快速溶出要求,这进一步说明现行标准ChP2020溶出条件区分力弱。
2.2.2 含量均匀度及含量测定
艾司唑仑片现行标准ChP2020 含量均匀度及含量测定采用UV 吸收系数法,文献[34]报道该方法受操作过程、仪器等因素影响较大,易造成测定结果的偏差。另外,杨惠霞等[35]采用紫外分光光度法测定艾司唑仑含量,发现片剂辅料于268 nm 波长下有紫外吸收,结果相对偏高,同时采用HPLC 法测定艾司唑仑含量,结果更准确。一致性评价后,质量标准及USP2022 均采用HPLC 法进行含量测定,虽然色谱条件各不相同,但提示HPLC 法能更准确地测定艾司唑仑含量。USP2022 艾司唑仑片与注册标准YBH08682021 和YBH06222021的色谱条件相同,均采用HPLC 外标法(等度洗脱),色谱柱采用填料L11柱(150 mm×4.6 mm,3µm),流动相为质量浓度为2.8 g/L 的磷酸二氢钾溶液(用1 mol/L氢氧化钠溶液调pH 至6.5)-甲醇-乙腈(55∶35∶10,V/V/V),检测波长为254 nm,供试品溶液质量浓度为0.02 mg/mL。质量标准YBH03582022 中的色谱条件同其有关物质的色谱条件。注册标准YBH09462021 中的色谱柱为C18柱,流动相为乙腈-水(40∶60,V/V),检测波长为223 nm,供试品与对照品溶液质量浓度均为10µg/mL。参照注册标准YBH09462021的色谱条件,建立测定艾司唑仑片含量的HPLC 法,发现未通过一致性评价产品UV 吸收系数法含量测定结果普遍高于HPLC法,2 种方法含量最大差值近10%。可见,受辅料及滤过方式的影响,会导致UV吸收系数法测定结果偏高。
3 问题与展望
目前,关于第二类精神药品艾司唑仑研究的国内外文献大多基于临床使用数据,而质量评价的报道较少。本研究中通过比较艾司唑仑原料药及其片剂国内外质量标准中关键质量属性指标控制差异,结合国产艾司唑仑原料药及其片剂生产工艺特点,发现目前国内艾司唑仑原料药及其片剂质量标准中存在可能引起安全性、有效性及均一性的风险。艾司唑仑原料药现行标准ChP2020主要存在有关物质缺失特定杂质控制、限度设置不合理的问题。国内原料药生产企业应结合自身合成工艺,开展基因毒性杂质与残留溶剂研究,基于风险评估制订合理控制策略,降低药物安全性风险。艾司唑仑制剂在溶出度、含量均匀度、含量测定等有效性方面,测定方法缺乏科学性;在有关物质控制方面,杂质控制及限度设置缺乏合理性,可能无法反映国产艾司唑仑片的质量差异。通过对比发现,国内艾司唑仑片通过一致性评价的注册标准整体提升,更有利于产品质量控制,但工艺杂质限度设置不合理。建议尚未通过一致性评价的生产企业在开展本品种质量研究时应重点关注有关物质、杂质谱、基因毒性杂质、溶出度、含量均匀度、含量测定等关键质量属性指标,优化产品处方及工艺设计,提高产品质量。同时,建议修订ChP2020艾司唑仑原料药及其片剂的质量标准,以满足已上市药品质量监管的需求,保障用药的安全性和有效性。
