高效液相色谱法测定枸橼酸芬太尼注射液有关物质含量*
2024-02-25宋文涛曾令高高梓真王佳瑜许向阳
宋文涛,曾令高,高梓真,王佳瑜,冯 旭,许向阳△
(1.江苏恩华药业股份有限公司药物研究院·江苏省中枢神经药物研究重点实验室,江苏 徐州 221000;2.重庆市食品药品检验检测研究院·国家药品监督管理局麻醉精神药品质量监测重点实验室,重庆 401121)
枸橼酸芬太尼注射液为强效镇痛药,适用于麻醉前、中、后期的镇静与镇痛,是目前复合全身麻醉中的常用药物,临床用法为静脉注射、硬膜外给药及静脉滴注[1-8],其风险远高于口服、肌肉注射等给药方式[9-10],故应严格控制产品质量。枸橼酸芬太尼注射液法定质量标准有《中国药典》(ChP)[11]、《美国药典》(USP)[12]和《英国药典》(BP)[13],但USP 中未对有关物质进行控制,ChP 中仅控制1 个特定杂质(杂质Ⅰ),BP 中控制2个特定杂质(杂质Ⅰ和杂质Ⅱ)。为进一步明确枸橼酸芬太尼注射液的潜在杂质,保证用药安全,本研究中建立了测定枸橼酸芬太尼注射液有关物质含量的高效液相色谱(HPLC)法,并进行了方法学验证,旨在提升现有枸橼酸芬太尼注射液的质量标准。现报道如下。
1 仪器与试药
1.1 仪器
Agilent 1260 型高效液相色谱仪(美国Agilent 公司);LC-20AD 型高效液相色谱仪(日本Shimadzu 公司);ME403T型电子分析天平(精度为1 mg),XSE205型电子分析天平(精度为10µg),XP6型电子分析天平(精度为1 µg),Seven Excellence 型多参数测定仪,均购自瑞士Mettler Toledo 公司;SHH-200GD-2 型药物强光照射试验箱(重庆永生实验仪器厂);DGG-9030B型电热恒温鼓风干燥箱(上海森信实验仪器有限公司)。
1.2 试药
枸橼酸芬太尼对照品(批号为171204-201905,含量为99.9%),杂质Ⅰ对照品(批号为171268-201602,含量为100.0%),均购自中国食品药品检定研究院;杂质Ⅱ对照品(批号为W986640,含量为99.6%),杂质Ⅲ对照品(批号为1030988,含量为98.8%),均购自英国LGC公司;杂质Ⅺ(上海阿拉丁生化科技股份有限公司,批号为D1912161,含量为99.107%);枸橼酸芬太尼注射液(江苏恩华药业股份有限公司,规格为每支10 mL∶0.5 mg,批号分别为ZA20211201,ZA20211202,ZA20 211203;规格为每支2 mL∶0.1 mg,批号分别为ZB20211201,ZB20211202,ZB20211203);枸橼酸、氯化铵均为分析纯,乙腈、甲醇均为色谱纯,水为超纯水。
2 方法与结果
2.1 色谱条件
色谱柱:Shim-pack scepter C18-120 柱(150 mm×4.6 mm,5µm);流动相:A 为0.01 mol/L 氯化铵溶液-乙腈(950∶50,V/V),B 为0.01 mol/L 氯化铵溶液-乙腈(400∶600,V/V),梯度洗脱(0~7 min时30%B →40%B,7~25 min 时40%B →100%B,25~40 min 时100%B,40~45 min 时100%B →30%B,45~60 min 时30%B);流速:1.0 mL/min;检测波长:210 nm;柱温:40 ℃;进样量:100µL。
2.2 溶液制备
空白溶液:取枸橼酸适量,加水溶解并定量稀释成每1 mL约含枸橼酸0.028 mg的溶液。
对照品溶液:取杂质Ⅰ、杂质Ⅱ、杂质Ⅲ和杂质Ⅺ对照品各适量,精密称定,加甲醇溶解,用水稀释成每1 mL约含各杂质5µg的溶液,即得对照品贮备液;取对照品贮备液适量,加水溶解,稀释成每1 mL约含各杂质0.25µg的溶液,即得。
系统适用性溶液:分别取枸橼酸芬太尼对照品和对照品贮备液各适量,加水溶解,稀释成每1 mL约含芬太尼50µg、各杂质0.25µg的溶液,即得。
供试品溶液:取枸橼酸芬太尼注射液适量,直接作为供试品溶液。
加样供试品溶液:取对照品贮备液0.5 mL,置10 mL容量瓶中,加供试品溶液定容,摇匀,即得
2.3 方法学考察
系统适用性试验:取2.2 项下系统适用性溶液适量,按2.1项下色谱条件进样测定,记录色谱图。结果芬太尼及各特定杂质的色谱峰均能达到基线分离,分离良好。详见图1。
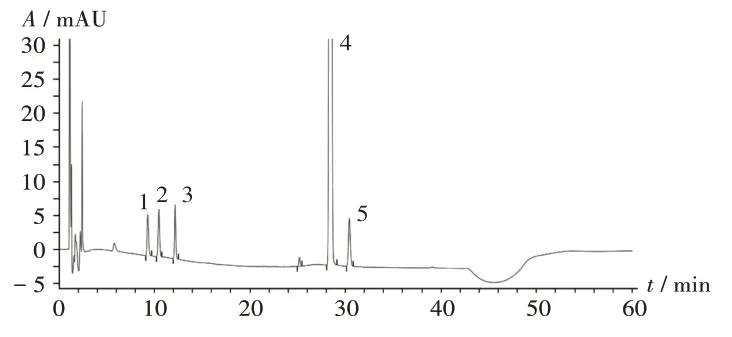
1.杂质Ⅲ 2.杂质Ⅺ 3.杂质Ⅱ 4.芬太尼 5.杂质Ⅰ图1 系统适用性试验高效液相色谱图1.Impurity Ⅲ 2.Impurity Ⅺ 3.Impurity Ⅱ 4.Fentanyl 5.Impurity ⅠFig.1 HPLC chromatogram of the system suitability test
破坏性试验:取2.2 项下供试品溶液适量,分别在酸(3 mol/L 盐酸溶液破坏24 h)、碱(3 mol/L 氢氧化钠溶液破坏24 h)、氧化(10%过氧化氢溶液破坏60 min)、高温(105 ℃破坏24 h)、光照[光照强度(5 000±500)lx,紫外光强度90µW/cm2,24 h]条件下进行破坏性试验,取破坏后的供试品溶液和未破坏的供试品溶液各适量,按2.1 项下色谱条件进样测定,破坏后的供试品溶液延长有机相比例最大洗脱时间段,运行时间延长至90 min,结果见图2。可见,拟订的有关物质测定方法对枸橼酸芬太尼注射液强制降解产生的杂质均可有效分离;氧化破坏条件下,杂质Ⅱ增加;酸破坏条件下,杂质Ⅰ略有增加;光照破坏条件下,在22~30 min 时会产生系列未知杂质,为芬太尼的体内代谢产物,故按非特定杂质进行控制。
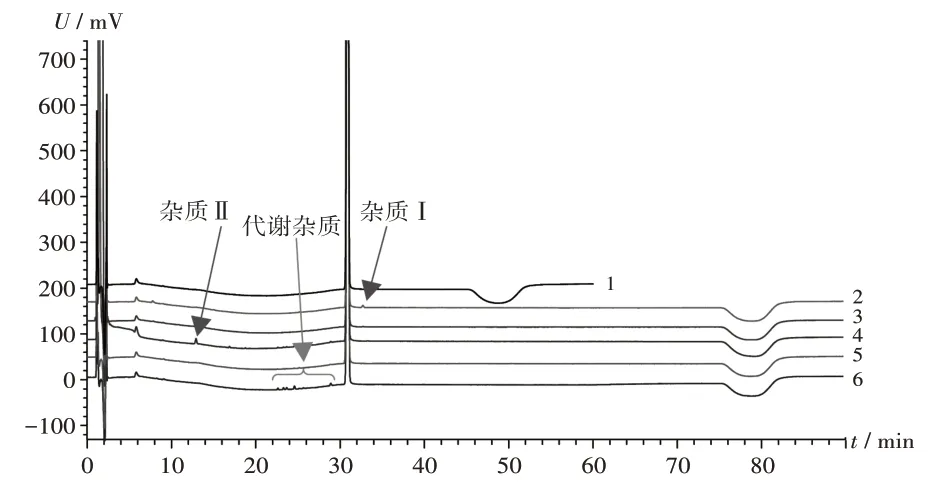
1.未破坏 2.酸破坏 3.碱破坏 4.氧化破坏 5.高温破坏 6.光照破坏图2 破坏性试验高效液相色谱图1.No destroying 2.Destroyed by acid 3.Destroyed by alkali 4.Destroyed by oxidation 5.Destroyed by high temperature 6.Destroyed by lightFig.2 HPLC chromatogram of the destructive test
线性关系考察:取枸橼酸芬太尼对照品和各杂质对照品各适量,精密称定,按2.2 项下方法制备系列浓度的标准曲线溶液,按2.1 项下色谱条件进样测定,以各成分质量浓度(C,µg/mL)为横坐标、峰面积(A)为纵坐标进行线性回归,并以主成分线性方程的斜率除以各杂质线性方程的斜率计算校正因子。结果见表1。
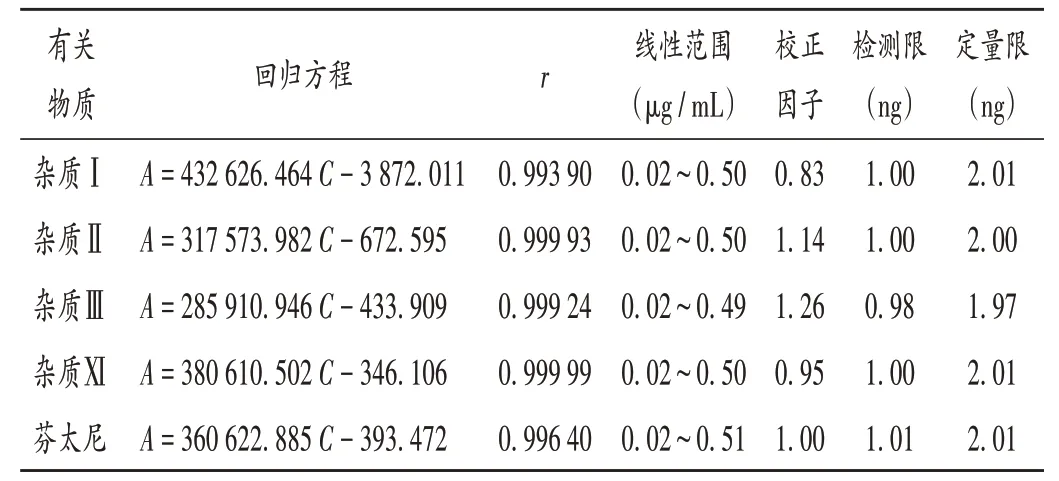
表1 线性关系考察、校正因子、检测限及定量限确定结果Tab.1 Results of the linear relation test,correction factor,LOD and LOQ
检测限与定量限确定:精密量取2.2项下对照品溶液适量,用水逐级稀释,以各杂质信噪比(S/N)≥3 时的样品量为检测限,以S/N≥10 且连续6 次进样峰面积的RSD≤10%时的样品量为定量限。结果见表1。
重复性试验:取2.2 项下加样供试品溶液6 份,按2.1 项下色谱条件进样测定。结果各杂质的RSD均小于2.0%(n=6),表明方法重复性良好。
中间精密度试验:精密量取2.2项下对照品溶液适量,按2.1项下色谱条件,由不同试验人员在不同日期采用不同品牌的高效液相色谱仪进样检测。结果各杂质的RSD均小于3.0%(n=12),表明方法中间精密度良好。
稳定性试验:精密量取2.2项下供试品溶液与对照品溶液各适量,分别于室温下放置0,9,18,27,36,48,60,72 h 时按2.1 项下色谱条件进样测定。结果供试品溶液与对照品溶液中各杂质及主峰面积的RSD均小于1.0%(n=8),表明供试品溶液与对照品溶液在室温下放置72 h内稳定性良好。
加样回收试验:精密量取2.2 项下对照品贮备液0.25,0.50,0.75 mL,置20 mL容量瓶中,加供试品溶液定容,分别作为50%,100%,150%浓度水平的加样回收试验溶液,平行3份,按2.1项下色谱条件进样测定。结果见表2。

表2 加样回收试验结果(n=9)Tab.2 Results of the recovery test(n=9)
耐用性试验:取2.2 项下系统适用性溶液、对照品溶液、加样供试品溶液各适量,分别在不同检测波长(208,210,212 nm)、柱温(38,40,42 ℃)、流速(0.9,1.0,1.1 mL/ min)、流动相盐溶液pH(9.4,9.6,9.8)、流动相比例及不同批次色谱柱条件下进样测定。结果不同条件下,主峰及各杂质的分离度均大于1.5,且有关物质含量无明显差异,表明方法耐用性较好。
2.4 样品有关物质含量测定
取样品(规格为每支10 mL∶0.5 mg,批号分别为ZA20211201,ZA20211202,ZA20 211203;规格为每支2 mL∶0.1 mg,批号分别为ZB20211201,ZB20211202,ZB20211203),按2.2 项下方法制备供试品溶液,按2.1项下色谱条件进样测定。按外标法以峰面积计算杂质Ⅺ含量,按外标法以对照品溶液中芬太尼校正后峰面积(分别乘以各杂质的校正因子)计算杂质Ⅰ、杂质Ⅱ及杂质Ⅲ的含量。结果6批样品中各特定杂质及未知杂质含量均小于0.05%(忽略限)。
2.5 方法验证
取2.2 项下系统适用性溶液,分别按2.1 项下色谱条件、ChP 色谱条件[11]、BP 色谱条件[13]进样测定。结果ChP 色谱条件下,杂质Ⅲ在溶剂峰位置出峰,且杂质Ⅰ与杂质Ⅱ未达到基线分离,色谱图见图3 A;BP 色谱条件下,杂质Ⅰ与杂质Ⅺ未达到基线分离,色谱图见图3 B。按2.1项下色谱条件进样测定,主峰及各杂质峰分离度均大于1.5,分离良好。

1.杂质Ⅲ 2.杂质Ⅺ 3.杂质Ⅱ 4.杂质Ⅰ 5.芬太尼 6.杂质Ⅰ和杂质ⅪA.ChP法 B.BP法图3 方法验证试验高效液相色谱图1.Impurity Ⅲ 2.Impurity Ⅺ 3.Impurity Ⅱ 4.Impurity Ⅰ 5.Fentanil 6.Impurity Ⅰ and impurity ⅪA.ChP method B.BP methodFig.3 HPLC chromatogram of the method validation test
3 讨论
3.1 制剂杂质谱分析
杂质Ⅰ[N-(1-苯乙基-4-哌啶基)苯胺]为枸橼酸芬太尼合成的中间体Ⅱ,ChP 标准及BP 标准均将其作为已知杂质进行控制;杂质Ⅱ(芬太尼氮氧化物)在氧化破坏条件下检出,为降解杂质;杂质Ⅲ(N-苯基-N-4-哌啶基丙酰胺)来源于枸橼酸芬太尼中间体Ⅱ的合成过程,中间体Ⅰ中残留的杂质哌啶酮和苯胺反应生成N-苯基哌啶-4-胺,在后续反应中和丙酸酐反应生成杂质Ⅲ,样品在光照破坏条件下亦略有检出;杂质Ⅺ(N-苯基丙酰胺)为枸橼酸芬太尼中间体Ⅱ中残留的苯胺和丙酸酐反应生成的产物,亦可由枸橼酸芬太尼降解产生。综合分析各杂质的来源及枸橼酸芬太尼注射液后续稳定性试验检出情况,最终将杂质Ⅰ、杂质Ⅱ和杂质Ⅺ作为特定杂质纳入枸橼酸芬太尼注射液的质量控制标准。
3.2 杂质限度确定
ChP 标准控制1 个特定杂质(杂质Ⅰ),限度为标示量的0.5%,最大未知杂质不得过0.25%,总未知杂质不得过0.75%;BP标准控制2个特定杂质(杂质Ⅰ和杂质Ⅱ),限度均为标示量的0.5%,最大未知杂质不得过0.25%,总未知杂质不得过0.75%。按各国药典标准中有关物质标准从严制订标准限度,结合2020 年版《中国药典(四部)》9101 分析方法验证指导原则[14]、人用药品技术要求国际协调理事会(ICH)Q3B R2 新药制剂中杂质[15]的要求,规定本品含杂质Ⅰ、杂质Ⅱ和杂质Ⅺ不得过标示量的0.5%,最大未知杂质不得过0.2%,总未知杂质不得过0.75%。
3.3 方法评价
本研究中建立的方法操作简便,专属性强,灵敏度、耐用性好,准确度、精密度较高,可用于枸橼酸芬太尼注射液中有关物质的含量测定。
