RDH12-associated retinal degeneration caused by a homozygous pathogenic variant of 146C>T and literature review
2024-02-23JinLiYiQunHuHongBoChengTingWangLongHaoKuangTaoHuangXiaoHeYan
Jin Li, Yi-Qun Hu, Hong-Bo Cheng, Ting Wang, Long-Hao Kuang, Tao Huang, Xiao-He Yan
Shenzhen Eye Hospital, Jinan University, Shenzhen Eye Institute, Shenzhen 518040, Guangdong Province, China
Abstract
● KEYWORDS: RDH12 gene; inherited retinal degeneration; homozygous pathogenic variant; clinical feature; multi-mode imaging
INTRODUCTION
Retinol dehydrogenase 12 (RDH12) is a nicotinamide adenine dinucleotide phosphate hydrogen (NADPH)-dependent retinaldehyde reductase that is specifically expressed in photoreceptor cells and participates in the visual cycle[1-2].In the visual cycle, a series of reactions are involved in the transport mechanisms between the retinal pigment epithelium (RPE) and photoreceptor cells, in which different members of retinol dehydrogenase play important roles[3-4].It has the highest activity with all-trans retinal production.In the presence of nicotinamide adenine dinucleotide phosphate(NADP) or NADPH as a cofactor, RDH12 acts on the inner segments of the cone and rod photoreceptor cells to catalyze the conversion of all-trans retinal into all-trans retinol[5-7].TheRDH12gene consists of 7 exons and encodes a protein with 316 amino acids.Gene variants inRDH12that interfere with the activity of this protein will cause a disruption of the visual cycle[8], leading to early and progressive retinopathy[9-10].RDH12variant-related retinal degeneration usually presents early vision loss in infancy or childhood[11].It is an autosomal recessive progressive retinal degenerative disease characterized by several types of retinopathy including Leber congenital amaurosis (LCA)[12-18]or/and early-onset severe retinal dystrophy (EOSRD)[13-14,16-17,19-20], autosomal recessive retinitis (RP)[13-14,19,21-22], cone-rod dystrophy(CORD)[13-14,19-20,23]and macular dystrophy[19-20], which leads to severe visual impairment or even blindness.Here we reported an unusual patient with predominant macular dystrophy caused by anRDH12homozygous pathogenic variant [c.146C>T(p.T49M)] and reviewed clinical characteristics of a series of reported cases with the same pathologic variant.
SUBJECTS AND METHODS
Ethical ApprovalThe Ethics Committee of Shenzhen Eye Hospital approved this study and the approved number is 20210603-11.The informed consent was obtained from the patient.
The patient underwent a complete ophthalmologic examination including best-corrected visual acuity assessment, anterior segment and dilated fundus examination, visual field test, spectral-domain optical coherence tomography and electroretinogram (ERG).We extracted genomic DNA from 2 mL peripheral blood and screened protein coding sequences of targeted retinal disease gene panels by chip capture highthroughput sequencing and confirmed by Sanger sequencing.DNA Extraction and Targeted Next Generation SequencingGenomic DNA (gDNA) was isolated using TIANamp Blood DNA kit (Tiangen, Shanghai, China) from ethylene diamine tetraacetic acid (EDTA) anticoagulated peripheral blood.Targeted next generation sequencing and annotation of variants were proceeded in Shanghai WeHealth Biomedical Technology Co, Ltd.(Shanghai, China).Briefly, genome libraries were constructed using the Hieff NGS OnePot DNA Library Prep Kit (YEASEN, Shanghai, China).Probes were synthesized by Integrated DNA Technologies, Inc.(Iowa, USA) to capture exons of 386 genes related to vitreoretinal diseases.Paired-end sequencing were performed by MGISEQ-T7 (BGI Genomics Co., Ltd., Shenzhen, China) to generate sequencing data of 150× depth.The quality of raw data was checked by FASTQC software.After removing the low-quality reads, the adaptor reads were mapped to the reference genome (GRCh37/hg19)with Burrows-Wheeler Aligner (BWA), Picard, and SAMtools.Genome Analysis Toolkit (GATK) was also used for insertion and deletion realignment, quality recalibration, and variant calling.Detected variants were annotated using ANNOVAR.Variants were filtered with minor allele frequencies, deleterious predictions by bioinformatic tools, gene functions and multiple databases.Then we reviewed the characteristics of the patients reported with the same homozygous variant.
RESULTS
A 30-year-old man came to our hospital for the first time,and he complained that he had night blindness since he was a child, and his vision began to decline at the age of 13, and worsen in recent 3y.He also had obvious vitreous floaters in the past 6y and occasionally metamorphopsia in the past 4y.Recently, he experienced transient vision loss in the last 4mo and both eyes suddenly darkens with dizziness.He has a family history of intermarriage that his grandfather and grandmother were siblings.The standard logarithm visual acuity chart was used to measure the visual acuity.The best corrected vision in his left and right eye was 0.04 and 0.12,respectively.The fundus photo showed bilateral macular atrophy, but larger areas of macular atrophy in the left eye.Punctate pigmentation was found at the posterior pole in the right eye, whereas few pigments around macula in the left eye.Autofluorescence showed roughly symmetrical hypofluorescence, especially in the macular area, the nasal side of the optic disc, the horizontal line between the optic disc and the macula, but in the temporal side of the optic disc it showed a crescent-shaped hyperfluorescence area.The peripheral retina is scattered with small patches of hypofluorescence,and streak-like hyperfluorescence appears in the border area of hypofluorescence (Figure 1C, 1D).Optical coherence tomography showed obvious macular atrophy, and the central macular thickness was significantly decreased in both eyes,but the severity of macular atrophy was inconsistent.The outer retina of the right eye was obviously thinned, the structure of each layer was blurred, and the RPE layer was atrophied with scattered hyper-reflective foci.The macular area of the left eye showed macularexcavation changes, with RPE layer rupture and the outer retina was markedly thinned.But relatively normal retinal structures were preserved in some areas between optic nerve head and macula (Figure 2).ERG revealed that the amplitudes of a- and b-wave were extinguished during photopic and scotopic conditions.Multifocal electroretinogram(mf-ERG) showed decreased amplitudes in the local macular area.Visual evoked potential showed significantly reduced amplitudes and 24-2 visual field tests showed severely central visual field defects in both eyes (Figure 2).Toxoplasma or other microorganisms infection tests were negative.We extracted genomic DNA from 2 mL peripheral blood and screened protein coding sequences of targeted retinal disease gene panels by chip capture high-throughput sequencing and confirmed by Sanger sequencing.A homozygous missense pathologic variant c.146C>T (chr14:68191267) was found,and the corresponding amino acid change was p.Thr49Met(Figure 3).In genomAD (Genome Aggregation Database), the frequency of this allele in all humans is 0.00003286, and it is 0.0003848 in east Asian populations.
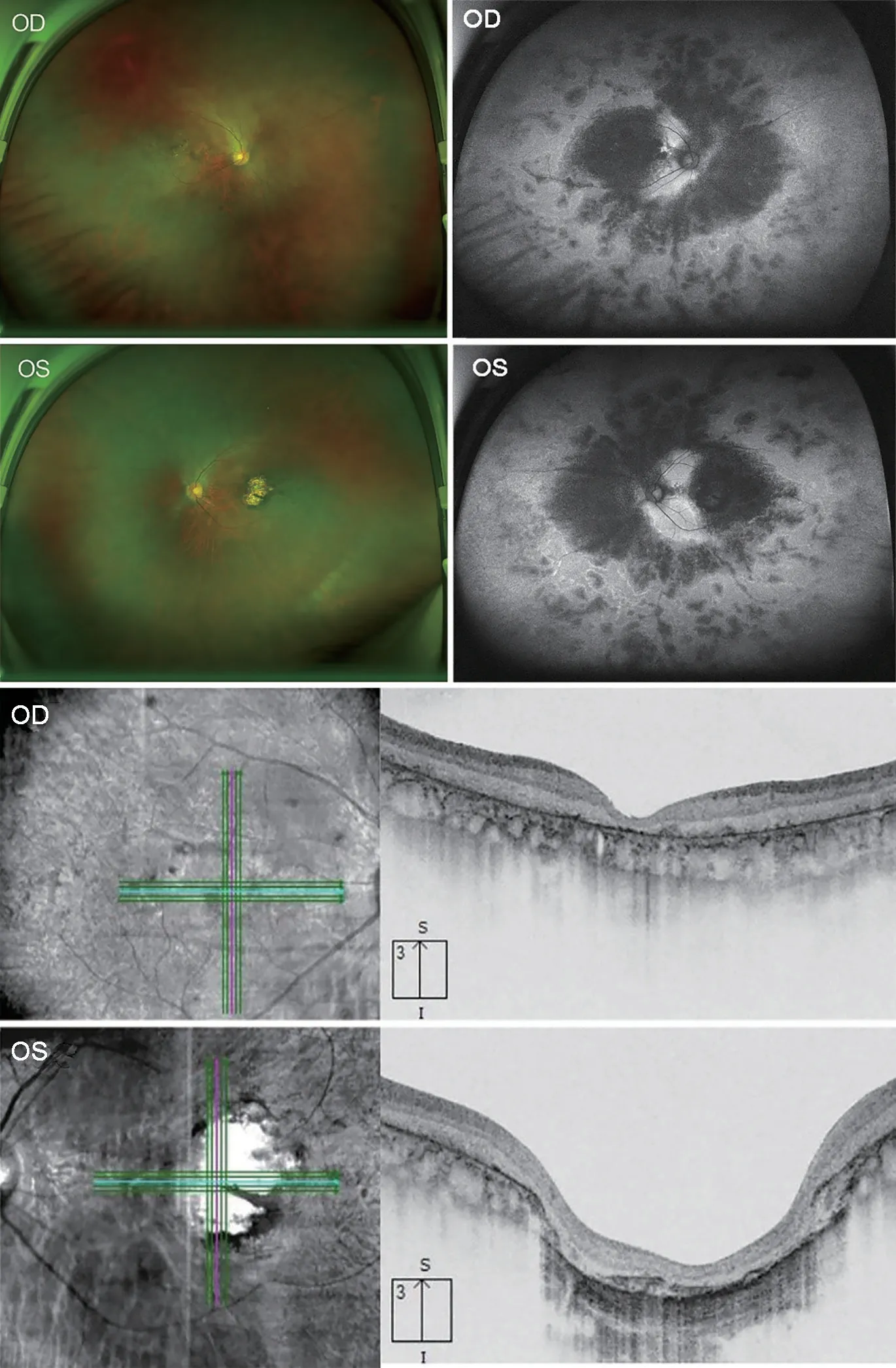
Figure 1 The fundus photo showed macular atrophy of both eyes,but the left eye was more pronounced Autofluorescence shows roughly symmetrical hypofluorescence, especially in the macular area, the nasal side of the optic disc, the horizontal line between the optic disc and the macula, but in the temporal side of the optic disc it showed a crescent-shaped hyperfluorescence area.The peripheral retina is scattered with small patches of hypofluorescence,and streak-like hyperfluorescence appears in the border area of hypofluorescence.Optical coherence tomography showed obvious macular atrophy, and the central macular thickness was significantly decreased in both eyes, but the severity of macular atrophy was inconsistent.The outer retina of the right eye was obviously thinned,the structure of each layer was blurred, and the RPE layer was atrophied with scattered hyper-reflective foci.The macular area of the left eye showed macular excavation changes, with RPE layer rupture and the outer retina was markedly thinned.But relatively normal retinal structures were preserved in some areas between optic nerve head and macula.RPE: Retinal pigment epithelium.
Then we reviewed the articles with the sameRDH12pathogenic variant from PubMed, GeenMedical and Embase and found a total of 3 articles including 12 patients withRDH12pathologic homozygous variant (c.146C>T) (Table 1).Based on these data and our case, this study summarizes general information, visual acuity, ERG, fundus phenotypic characteristics, and the diagnosis of the 13 patients, as shown in Table 1.We concluded that 6 patients were female, 6 patients were male, and 1 patients had unrecorded gender.The age distribution ranged from 4 to 30 years old, and all had visual impairment.Among them, 5 patients had best corrected visual acuity equal to or less than 0.05 (convert to new national standard visual acuity chart) in one or both eyes.But in two patients, the vision acuity was not available.The onset age of disease was recorded in 6 patients, most of whom were younger than 10 years of age, and whose initial symptoms included night blindness, visual impairment, and poor school performance.The ocular examination included fundus photography and ERG.The ERG showed decreased amplitudes in 6 patients, and extinguished waveform in the remaining 7 patients[1,23-25].However, the age of onset and clinical manifestations of these patients were different, and the decline in visual acuity varied, and the pathological morphological changes of their posterior retina and macular area were also different.Among the other 12 patients, 7 patients had fundus examination records, including one with typical RP and five with bilateral coloboma-like atrophic macular chorioretinal scars and scattered areas of RPE hyperplasia.Although there are the same variants among the patients, there are different clinical diagnoses, including CORD (6/13), LCA (5/13) and autosomal recessive RP (1/13), and our patient was diagnosed with late-onset retinal degeneration.In patients with same family (No.2-7 in one family; No.8-12 in one family), they have the same retinal phenotypes.
DISCUSSION
Genetic testing is usually used to identify the potential cause of congeneital eye diseases[26].RDH12-relatedretinopathy should also be confirmed by genetic testing.It is characterized by severely impaired visual function in early life, which begins in 2 to 4y.The vision gradually deteriorated and most patients had significant vision loss before the age of 30[25].The most severe condition may be obvious visual impairment in the first six months of life[13].There are different clinical diagnoses in the reported cases withRDH12pathologic variants, including EOSRD/LCA, CORD, RP, and macular dystrophy.InRDH12-related retinopathy, there are four types of pathological retinal morphological changes, including macular coloboma (mostly petal-like) with dense bone spicule pigmentation in the midperipheral retina, macular discoloration and wide-spread bone spicule and/or salt-pepper pigmentation, heavy and confluent pigment proliferation in the mid-peripheral region involving the macular region, and retinal posterior pole atrophy with a relatively normal peripheral retina[14].In our patient, macular dystrophy was found similar to the reported cases, but the it is still different from the four retinal phenotypes.Specifically,macular atrophy with sparse pigmentation was found in both eyes but without dense bone spicule pigmentation.The diameter of the retinal arteries and veins on the nasal side of both eyes is significantly thinner, but there was no obvious abnormality in the mid-peripheral part of the temporal retinal vessels, which is associated with the preservation of crescentshaped hyperfluorescence in the temporal side from fundus autofluorescence (FAF) images.Peripapillary sparing was also found in the patient, which was also reported in another case[27].The pattern of macular dystrophy between two eyes are not consistent.
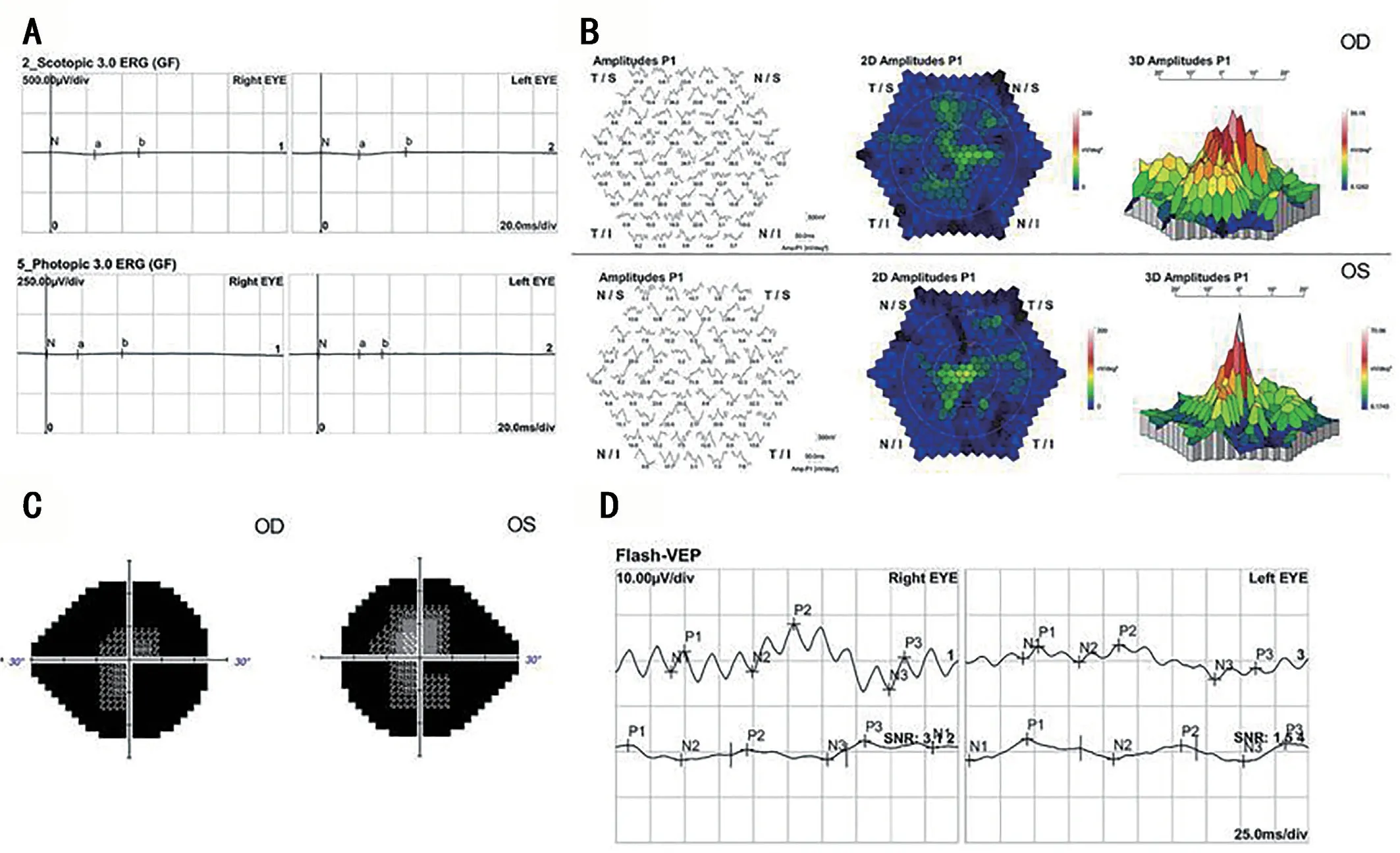
Figure 2 Visual functional changes in the patient Electroretinogram (ERG) revealed that the amplitudes of a- and b-wave were extinguished during photopic and scotopic conditions (A); Multifocal electroretinogram (mf-ERG) showed abnormal changes in the local function of the macular area (B).Visual field showed that the patient only had a very limited central field, and the peripheral field disappeared (C) and visual evoked potential showed significantly reduced amplitudes (D).
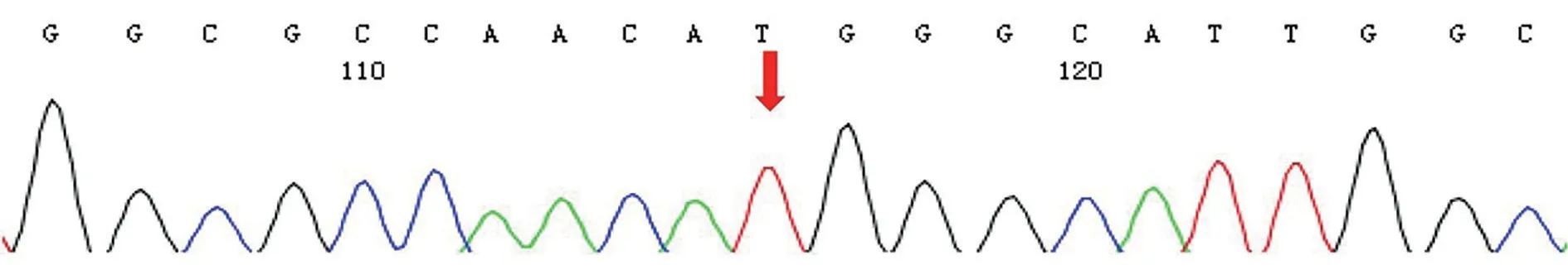
Figure 3 A homozygous missense pathologic variant c.146C>T in RDH12 in the patient.
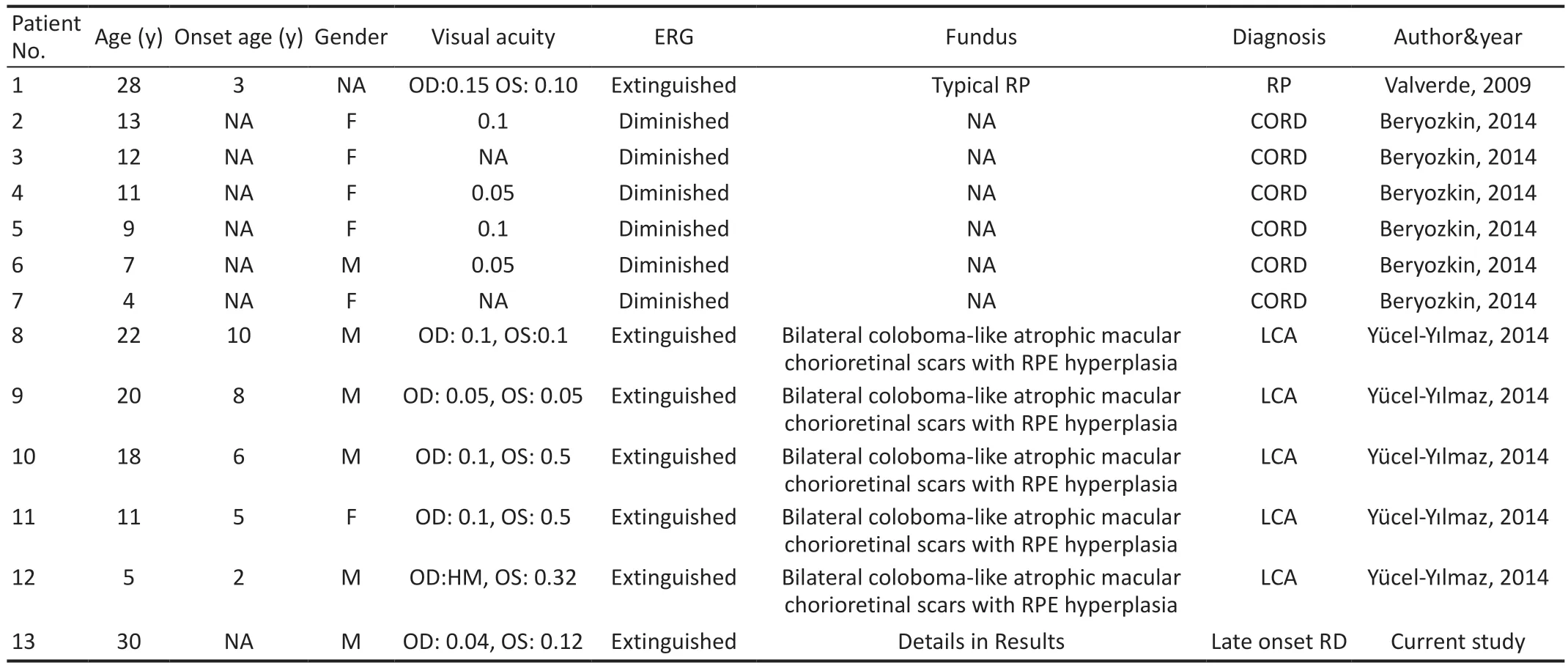
Table 1 Clinical features of RDH12-induced retinal degeneration caused by the mutation of 146C>T (p.T49M) from the published literatures
In addition, the progression of our patient’s visual function also differs from other patients reported with the same variant.In our study, the patient had night blindness since childhood, but his vision declined later until 13 years old.At the age of 18,he gave up the driver’s license test since his corrected visual acuity was only 0.5 in both eyes.In the past 20y, the visual acuity has been gradually declining, and his visual acuity is severely reduced now.However, a previous study showed that thatRDH12T49M substitution is related to an overall better visual acuity and milder phenotype[28].In this study,most patients were younger, and the mean age is 18y, so the severity of retinal degeneration could be relatively milder in these patients.Another study also supported this point of more severe visual acuity decline in older age compared to younger age based on two patients with the homozygous variants[28].The homozygous variant of 146C>T (p.T49M) seems to be the most frequent homozygous genotypes (24/252) inRDH12[13].But in another study included 57 patients withRDH12variants,only two patients have the same homozygous variants[16].Therefore, it is still unclear if the homozygous variant of 146C>T (p.T49M) is a hot mutation ofRDH12.Compared with the clinical symptoms of 18 other reported patients, this patient showed later retinal degeneration than other patients.It is manifested as night blindness from an early age, and a significant decrease in vision occurs during adolescence, and the vision is poorly corrected.After that, it declined slowly for a long time, until it accelerated three years ago.Compared with the reported cases of poor vision since childhood, this has a longer course of disease progression.In this patient, he has a family history of intermarriage between his father and mother.The variant could be due to a geographic isolate.Patients with the same pathogenic variant may show different clinical phenotypes, and the phenotypic differences may be due to different genetic background, gene-environment interaction or possible presence of additional pathogenic variants in different genes.From the Table 1, several patients are from the same family and they had a common genetic background, but they are from different places and the differences in clinical manifestations may be due to geographic isolation.
At present, a suitable animal model ofRDH12-related retinal disease is not available, as there are very weak retinal phenotypes inRdh12knockout mice.Therefore, there are still huge challenges to develop novel treatment forRDH12-related retinopathy.In the future,RDH12-knockout monkeys should be tried to develop new animal disease model of the disease.It has been reported that adeno-associated virus (AVV)-mediatedRDH12gene replacement therapy has promising results in mice.HumanRDH12cDNA packaged in AAV2/5 vector was delivered toRdh12knockout mice by subretinal injection,resulting in correct expression positioning and maintain the structural integrity of the retina.Retinal homogenates from the injected knockout mice showed increased all-trans retinol formation and reduced sensitivity to light damage, but without visual cycle disruption[14-15].This study shows promise for future gene therapy of this disease.In addition, there have been many recent studies on new treatments for retinal damage,such as studies on the protection of the retina by human umbilical cord mesenchymal stem cells modified by the leukemia inhibitory factor, which have provided us with some inspiration for further treatment[29].A large number of previous studies have shown that the application of international, multi-center, retrospective cohort studies and accurate genetic testing technology will bring new insights into the construction of genotype-phenotype correlation databases and retinal treatments[30-31].
In summary, we reported an unusual patient with a homozygous pathogenic variant in the c.146C>T ofRDH12who presents late-onset and asymmetric retinal degeneration.However, among the reported patients, the age of onset,clinical characteristics and the rate of disease progression vary.The clinical manifestations of our patient with a comprehensive ophthalmological examination have increased the overall knowledge of this rare disease.
ACKNOWLEDGEMENTS
Authors’ contributions:Conceived and designed the experiments: Yan XH; Performed the experiments: Li J, Hu YQ, Wang T, Kuang LH, Huang T and Yan XH; Analyzed the data: Li J, Hu YQ, Cheng HB, Yan XH; Contributed reagents/material/analysis tools: Yan XH, Cheng HB; Wrote the manuscript: Li J, Yan XH; All authors reviewed the manuscript.
Foundations:Supported by Shenzhen Science and Technology Program, Shenzhen, China (No.JCYJ20200109145001814;No.SGDX20211123120001001); the National Natural Science Foundation of China (No.81970790); Sanming Project of Medicine in Shenzhen (No.SZSM202011015).
Conflicts of Interest:Li J,None;Hu YQ,None;Cheng HB,None;Wang T,None;Kuang LH,None;Huang T,None;Yan XH,None.
杂志排行

International Journal of Ophthalmology的其它文章
- Using choroidal thickness to detect myopic macular degeneration
- lmpact of multifocal gas-permeable lens designs on short-term choroidal response, axial length, and retinal defocus profile
- Baerveldt glaucoma implant with Supramid© ripcord stent in neovascular glaucoma: a case series
- Efficacy and safety of Usights UC100 illuminated microcatheter in microcatheter-assisted trabeculotomy
- Quantifying peripapillary vessel density and retinal nerve fibre layer in type 1 diabetic children without clinically detectable retinopathy using OCTA
- Nomogram to predict severe retinopathy of prematurity in Southeast China