Mo掺杂CoSe2纳米片阵列的制备及电催化性质研究
2024-02-20金舒婷
曹 健,金舒婷,张 炜,冯 博
(1.吉林师范大学 功能材料物理与化学教育部重点实验室,吉林 长春 130103;2.吉林师范大学 信息技术学院,吉林 四平 136000)
0 引言
电催化水裂解是制备高纯度氢的一种重要方法[1].全解水反应包括阴极的析氢反应(HER)和阳极的析氧反应(OER)[2].但由于在大电流下HER/OER的H*/O*吸附/解吸过程具有较大的能量势垒和传质电阻,因此急需一种高效的电催化剂降低过电位,加速反应过程[3].金属有机骨架(MOFs)由于其独特的结构、较大的比表面积和孔隙率被认为是合成催化剂的理想前驱体[4-6].过渡金属硒化物是常用的HER/OER催化剂.由于硒元素的3d轨道能量接近于3s和3p的轨道能量,因此它可以与过渡金属原子结合形成共价键.这种电子结构使过渡金属硒化物具有独特的金属性质,有利于电子的转移和反应[7].此外,过渡金属硒化物具有良好的催化活性和稳定性.CoSe2作为一种具有代表性的过渡金属硒化物,在电催化方面得到了广泛的研究[8].但CoSe2的离子扩散效率低,在电化学反应过程中体积膨胀大,严重阻碍了其大规模应用[9].为了解决上述问题,可以通过元素掺杂调控CoSe2形貌,增加边缘活性位点,调节CoSe2的电子结构和中间结合能.目前,常在CoSe2中掺杂Mo、Fe、Mn等金属元素提高HER、OER和全水解的催化活性[10-11].其中Mo掺杂可以提高材料的电导率和电子转移能力,同时还可以调节H*的吸附能,从而降低反应过程的能垒[12-13].
1 实验
1.1 样品制备
1.1.1 碳布的预处理
利用丙酮、去离子水和乙醇分别对碳布(20 mm×30 mm)超声20 min以除去表面的杂质,60 ℃干燥6 h.
1.1.2 ZIF-67@CC的合成
将0.582 g Co(NO3)2·6H2O(溶液A)和1.314 g 2-甲基咪唑(溶液B)分别分散在40 mL去离子水中,搅拌1 h使其完全溶解.随后将溶液B迅速倒入溶液A中形成溶液C.将已清洁的碳布浸入溶液C中静置4 h后,得到紫色的ZIF-67@CC,利用去离子水和乙醇清洗3次,并在60 ℃下真空干燥6 h.
1.1.3 Co LDH@CC的合成
将ZIF-67@CC放入100 mL去离子水和乙醇(V去离子水∶V乙醇=1∶9)的混合溶液中,在90 ℃下保持2 h.待到冷却至室温后取出样品,然后用去离子水和无水乙醇清洗,在60 ℃下真空干燥6 h.
1.1.4 CoSe2@CC的合成
首先,将2 mmol的Se粉末溶解在5 mL水合肼中(溶液A)、xmmol的Na2MoO4·6H2O(x=0.1、0.3、0.5、0.7、0.9)分散在10 mL去离子水中(溶液B),搅拌1 h使其完全溶解.然后,将溶液A倒入溶液B中继续搅拌2 h,将搅拌后的溶液和Co LDH@CC一起置于25 mL不锈钢反应釜中,在干燥箱中180 ℃加热12 h.最后,待高压釜冷却至室温后,用乙醇和去离子水洗涤样品,在60 ℃下真空干燥6 h.将Na2MoO4·6H2O用量为0.1、0.3、0.5、0.7、0.9 mmol的样品分别记为Mo-0.1、Mo-0.3、Mo-0.5、Mo-0.7、Mo-0.9.
1.2 样品表征
利用X射线衍射仪(XRD,MXP18,MAC Science)、扫描电子显微镜(SEM,JSM-7800F,JEOL)、X射线光电子能谱仪(XPS,ESCALAB 250,Thermo Scientific)等对样品进行测试表征.
1.3 电催化实验
在CHI 760e电化学工作站上使用标准三电极系统进行电催化测试,其中石墨作为对电极,Hg/HgO用作参比电极,样品(0.3×0.3 cm2)作为工作电极.在双电极电池系统中,样品既作为阴极又作为阳极进行全水解.所有电化学测试中使用的电解液均为1.0 mol/L KOH(pH=13.6).在进行电化学实验之前,利用N2通气30 min除氧.利用以下公式将电势转换为相对于可逆氢电极(RHE)的电势:
ERHE=ESCE+0.098+0.059×pH.
在1 mV/s的扫描速率下测试LSV曲线,该极化曲线在几个循环后达到稳态,所有测得的极化曲线均经过iR校正.为了进行比较,利用商用Pt/C或IrO2作为工作电极测试其电催化性能.稳定性通过计时电流响应来测量.在10-1~104Hz的频率范围内进行电化学阻抗谱(EIS)的测量.
2 结果与讨论
如图1所示,通过XRD对所制备样品的晶体结构进行表征.图1中位于26°的强衍射峰为碳布的(002)面衍射峰.位于30.7°、34.5°、35.9°、40.4°、47.7°、50.6°的衍射峰,分别对应CoSe2(JCPDS Card No.53-0049)的(101)、(111)、(120)、(211)、(130)、(122)的晶面[14].与CoSe2的晶体结构相比,Mo-0.3样品中没有杂质相关的衍射峰,说明Mo被成功掺杂到CoSe2的晶格中.Mo替代Co的位置将会导致CoSe2晶格缺陷或者电子重新排布,这将会提高CoSe2的催化活性.但是随着Mo元素掺杂量的增加,Mo-0.5样品的结晶性降低.

图1 CoSe2、Mo-0.3和Mo-0.5的XRD谱图Fig.1 XRD patterns of CoSe2,Mo-0.3 and Mo-0.5
如图2是Co-LDH、Mo-0.3和Mo-0.5的SEM图.在图2(A)中,Co-LDH纳米片在碳布上垂直且均匀的分布,表面粗糙.图2(B)为掺杂0.3 mmol Mo的扫描图,Mo-0.3在碳布上垂直排列且相互交错,构成片状阵列结构,同时没有明显的团聚现象.这种纳米片结构可以使得催化剂与电解液充分接触,二维片状结构可以暴露更多的催化活性位点,提供更多的反应中心,有利于电子的传输.另外纳米片阵列与碳布之间的强附着力有利于电子从电极表面转移到催化活性位点.但是从图2(C)看出,Mo-0.5样品的纳米片表面更加粗糙,彼此堆叠在一起,这不利于电催化过程中的离子传输.

图2 Co-LDH(A)、Mo-0.3(B)和Mo-0.5(C)的SEM图
图3是Mo-0.3的mapping图和EDS图.从图3(B)—(D)可以看出 Mo、Co和Se元素均匀地分布在Mo-0.3的表面.图3(E)证实了Mo元素成功掺杂进入CoSe2晶格中,掺杂的Mo原子比例为3.23%.

图3 Mo-0.3的mapping(A—D)和EDS(E)谱图
采用X射线光电子能谱(XPS)分析了样品的化学价态和组成.从图4(A)中可以看到样品中存在Mo、Co、Se、C和O元素(其中C和O元素可能来源于催化剂表面存在的H2O、O2、CO2).图4(B)—(D)分别是CoSe2和Mo-0.3的Co 2p、Se 3d、Mo 3d的XPS谱图.在Co 2p的XPS图中(图4(B)),778.8 eV和793.8 eV处的峰分别对应于Co 2p3/2和Co 2p1/2[15].在Se 3d的XPS图中(图4(C)),Se 3d5/2和Se 3d3/2的结合能分别为54.1、54.9 eV,对应于CoSe2中Se的化合价[16-17].此外,在Mo-0.3的Mo 3dXPS图中(图4(D)),位于228.4、231.6、232.9 eV处的衍射峰分别对应于Mo4+的Mo 3d5/2、Mo 3d3/2,说明Mo4+成功掺杂到CoSe2晶格中[18-19].位于236.0 eV处的峰则与Mo6+有关,是由于催化剂表面的轻微氧化所导致[20].掺杂Mo元素后,Co 2p、Se 3d的结合能变大,表明Co与Se之间的相互作用增强.这些结果进一步证明Mo取代Co掺入CoSe2的晶格中,导致CoSe2产生晶格缺陷且电子重新分布,这有助于提高Mo-CoSe2的催化活性.

图4 CoSe2和Mo-0.3的XPS全谱图(A)以及Co 2p、Se 3d和Mo 3d的XPS谱图(B—D)
为了评估不同Mo掺杂CoSe2样品的电化学性能,在1 mol/L KOH电解液当中进行了LSV、CV、EIS和计时电位测试.根据CV曲线绘制出双电容曲线(Cdl),如图5所示.Mo-0.3的Cdl值为11.74 mF/cm2,高于Mo-0.1 (4.35 mF/cm2)、Mo-0.5 (5.32 mF/cm2)、Mo-0.7 (2.80 mF/cm2)和Mo-0.9 (0.66 mF/cm2),说明Mo-0.3的电化学比表面积最大.
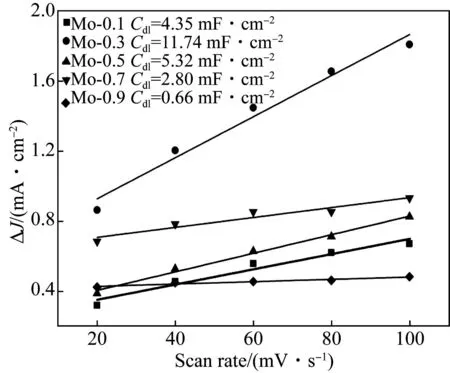
图5 Mo-0.1、Mo-0.3、Mo-0.5、Mo-0.7和Mo-0.9的双层电容Fig.5 The double-layer capacitance of Mo-0.1, Mo-0.3,Mo-0.5,Mo-0.7 and Mo-0.9
采用线性扫描伏安法(LSV),在氮气饱和的1 mol/L KOH溶液中对催化剂的HER性能进行测试.图6(A)为商用Pt/C、CoSe2、Mo-0.1、Mo-0.3、Mo-0.5、Mo-0.7和Mo-0.9样品在扫描速率为5 mV/s时测量的LSV曲线,并进行iR校正.除Pt/C外,Mo-0.3表现出优异的电化学性能,电流密度10 mA/cm2时,过电势为135 mV,明显低于其他催化剂.这是因为适量的Mo元素掺杂会提供更多的催化位点,片状结构会提供较大的比表面积.图6(B)是不同催化剂的Tafel斜率图.除Pt/C(28.75 mV/dec)外,Mo-0.3材料具有最小的Tafel斜率(54.90 mV/dec),Tafel斜率越小,说明其电化学反应速度最快.采用电化学阻抗谱进行测试,Mo-0.3具有较低的电荷转移电阻,较好的导电性,因此电催化反应速率较快.此外,在10 mA/cm2的电流密度下对Mo-0.3进行了60 h的稳定性测试(图6(D)).在连续测试60 h后,电流密度没有下降,1 000圈循环后的LSV测试结果较稳定,表明Mo-0.3催化剂具有优异的HER稳定性.
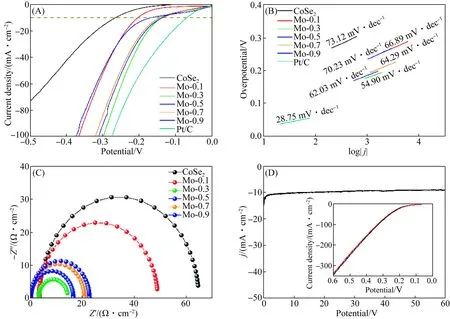
图6 CoSe2和Mo掺杂CoSe2样品的HER线性扫描伏安曲线图(A)、析氢性能的Tafel斜率图(B)、交流阻抗图(C)和Mo-0.3的计时电位曲线(插图为其循环1 000圈前后的HER曲线)(D)
图7为IrO2、CoSe2和Mo掺杂CoSe2催化剂的OER催化性能.图7(A)—(B)分别是在1 mol/L KOH水溶液中以5 mV/s的扫描速率进行的LSV测试以及Tafel图.结果表明Mo的掺杂会提高CoSe2的OER催化性能,其中Mo-0.3催化剂表现出了最好的OER性质,在电流密度为50 mA/cm2的情况下,过电势仅为350 mV,并且具有最小的Tafel斜率(88.05 mV/dec),证明具有较快的析氧反应速率.如图7(C)所示,Mo-0.3催化剂的Rct显著降低,表明Mo-0.3催化剂的电荷转移率显著提高,可促进电荷的转移.对Mo-0.3催化剂进行稳定性测试,在10 mA/cm2的电流密度下进行了60 h的稳定性测试(图7(D)).在连续运行60 h后,样品具有良好的稳定性,同时比较第1圈和第1 000圈循环的LSV测试曲线也无较大差异,表明Mo-0.3催化剂具有优异的OER稳定性.
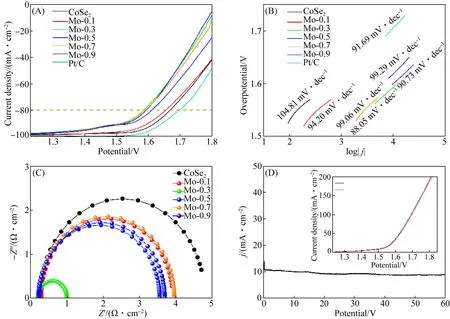
图7 CoSe2和Mo掺杂CoSe2样品的OER性能线性扫描伏安曲线图(A)、析氧性能的Tafel图(B)、交流阻抗图(C)以及Mo-0.3的计时电位曲线(插图为Mo-0.3循环1 000圈后的OER曲线)(D)
由于Mo-0.3催化剂具有良好的HER和OER活性以及稳定性,进一步利用Mo-0.3样品进行全水解测试.为了对比还组装了CoSe2(+)||CoSe2(-)和IrO2(+)||Pt/C(-)电催化剂.图8(A)为双电极系统在碱性电解液中全水解的极化曲线,当所输出的电流密度为10 mA/cm2时,电压仅为1.5 V,其催化效率高于贵金属基催化剂材料(IrO2(+)||Pt/C(-)).此外Mo-0.3催化剂在连续运行60 h后稳定性较好(图8(B)).
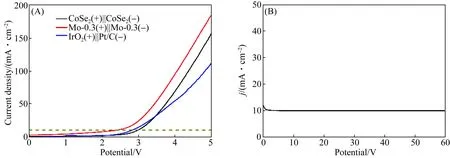
图8 CoSe2(+)||CoSe2(-),IrO2(+)||Pt/C(-),Mo-0.3(+)||Mo-0.3(-)的全解水性能线性扫描伏安曲线图(A)和Mo-0.3(+)||Mo-0.3(-)的计时电位曲线(B)
3 结论
在碳布上利用ZIF-67为前驱体制备了不同Mo掺杂量的CoSe2纳米片阵列.由于该样品具有良好的纳米片阵列形貌,加之引入适量的Mo掺杂发现可增加其活性位点,提高其导电性.其中Mo-0.3纳米片阵列表现出最佳的HER催化活性和OER催化活性.同时利用Mo-0.3催化剂作为碱性电解槽的阴极和阳极,在1.5 V的电压下就可以驱动10 mA/cm2的电流密度,并可连续产生氢气和氧气60 h.
