高效液相色谱法测定诃子中诃黎勒酸和诃子酸
2024-02-20王巍张强杨武杰郝季陈九妹安悦言鞠成国窦德强
王巍,张强,杨武杰,郝季,陈九妹,安悦言,鞠成国,2,窦德强
[1.辽宁中医药大学药学院,辽宁大连 116600; 2.国家中医药管理局中药炮制技术传承基地(辽宁),辽宁大连 116600]
诃子为使君子科植物诃子(Terminalia chebula Retz.)或绒毛诃子(T.chebula Retz.var.tomentella Kurt.)的干燥成熟果实,为《中华人民共和国药典》(以下简称《中国药典》) 2020 年版一部收载。诃子味苦、酸、涩,性平,归肺、大肠经,具有涩肠止泻、敛肺止咳、降火利咽的功效[1]。诃子化学成分主要为鞣质类[2],其中以诃黎勒酸、诃子酸含量较高[3-5]。国内外多项研究表明,诃黎勒酸、诃子酸具有良好的抗氧化、抗炎、抗肿瘤以及降血糖的作用[6-9]。赵鹿等[2]从植物亲缘学及化学成分特有性、传统功效、药性、药动学、新临床用途和化学成分可测性等多个方面预测包括诃黎勒酸、诃子酸在内的4 个化学成分为诃子的质量标志物。《中国药典》中诃子的质量标准缺少含量测定的要求,这对诃子药材的质量控制造成了一定的缺憾。同时现有文献也未见对诃黎勒酸、诃子酸同时进行含量测定的报道,多是对其它鞣质类成分进行测定[10-11]。笔者建立了测定诃子药材中诃黎勒酸、诃子酸含量的高效液相色谱法,作为《中国药典》2020年版一部中诃子质量标准的补充。
1 实验部分
1.1 主要仪器与试剂
高效液相色谱仪:LC-16型,日本岛津公司。
电子天平:FA1004B 型,感量为0.1 mg,上海精密科学仪器有限公司。
电子天平:METTLER AE240 型,感量为0.01 mg,瑞士梅特勒-托利多仪器有限公司。
医用超声波清洗器:KQ-250E 型,江苏昆山市超声仪器有限公司。
诃黎勒酸、诃子酸对照品:经HPLC法[12-13]检测,纯度(质量分数)不小于98%,自制。
乙腈:色谱纯,美国天地试剂公司。
甲醇、甲酸:均为色谱纯,天津科密欧化学试剂有限公司。
诃子药材:市售,产地为云南,经辽宁中医药大学药学院宋慧鹏副教授鉴定为使君子科植物诃子Terminalia chebula Retz.的干燥成熟果实。
实验用水为娃哈哈纯净水。
1.2 色谱条件
色谱柱:Dikma Platisil C18柱(250 mm×4.6 mm,5 μm,北京迪科马科技有限公司);柱温:30 ℃;检测波长:280 nm;流动相:体积分数为0.1%的甲酸水溶液-乙腈-甲醇(体积比为78∶16∶6),等度洗脱,流量为1 mL/min;进样体积:5 μL。
1.3 溶液制备
混合对照品溶液:诃黎勒酸、诃子酸质量浓度分别为0.410、0.455 mg/mL。精密称取诃黎勒酸、诃子酸对照品适量,加入50%(体积分数,下同)乙腈溶液溶解,并稀释、定容。
样品溶液:取诃子药材,去核,留果肉,粉碎,过孔径为250 μm (65目)筛。精密称取0.1 g,精密加入50%乙腈溶液25 mL,称定质量,超声(功率250 W,频率40 kHz)提取30 min,冷却至室温,以提取溶剂补足损失质量,摇匀,滤过。精密量取续滤液2 mL至10 mL容量瓶中,用50%乙腈溶液定容至标线,摇匀,再经0.45 μm 微孔滤膜滤过,取续滤液,作为样品溶液。
1.4 实验方法
取混合对照品溶液、样品溶液,按照1.2 色谱条件进样分析,记录色谱峰面积,按照外标法计算诃子药材中诃黎勒酸、诃子酸的含量。
2 结果与讨论
2.1 提取方式选择
取诃子药材,去核,留果肉,粉碎,过孔径为250 μm 筛。精密称取2 份,每份0.1 g,精密加入50%乙腈溶液25 mL,称定质量,分别超声(功率250 W,频率40 kHz)提取30 min、加热回流30 min;放冷,以提取溶剂补足损失质量,摇匀,滤过,精密量取续滤液2 mL至10 mL容量瓶中,用50%乙腈溶液定容至标线,摇匀,再经0.45 μm 微孔滤膜滤过,取续滤液进行测定。超声提取样品测得诃黎勒酸质量分数为13.95%,诃子酸质量分数为9.30%;加热回流30 min提取样品测得诃黎勒酸质量分数为13.27%,诃子酸质量分数为9.15%。说明超声提取效果更佳,因此提取方式应以超声提取为宜。
2.2 提取溶剂选择
取诃子药材,去核,留果肉,粉碎,过孔径为250 μm 筛。精密称取21 份,每份0.1 g,分别精密加入25 mL 的水、不同浓度的甲醇溶液或不同浓度的乙腈溶液,其余步骤同上,结果见表1。
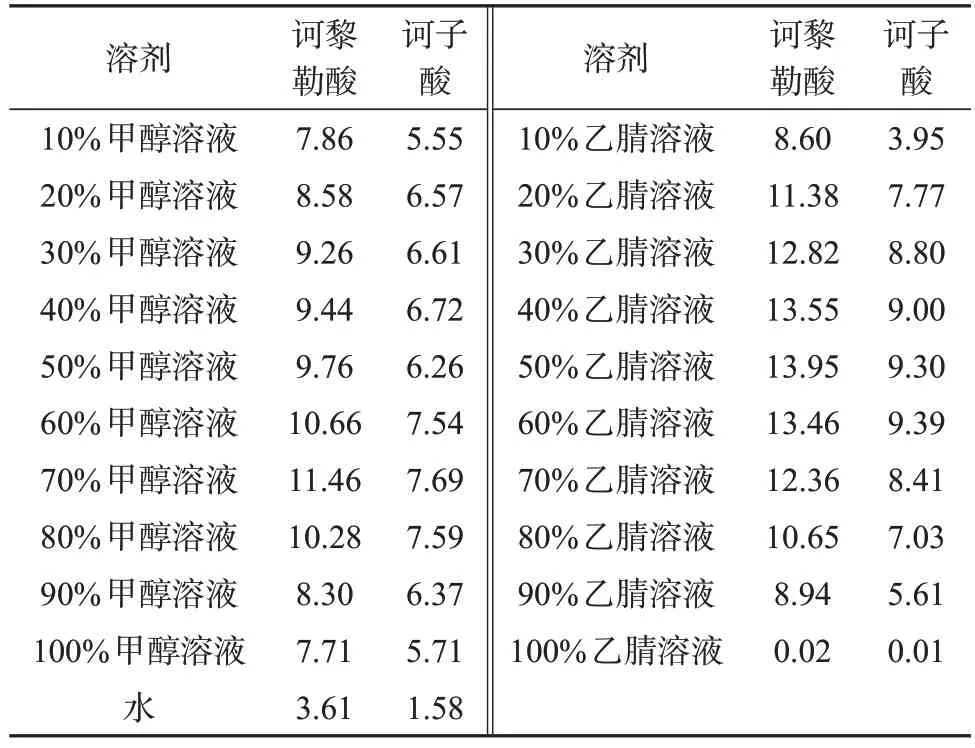
表1 不同提取溶剂时诃黎勒酸和诃子酸质量分数测定结果Tab.1 Determination results of mass fractions of chebulagic acid and chebulinic acid in different extraction solvents %
由表1可知,乙腈的提取率高于甲醇,其中50%乙腈溶液对两种成分的提取量均较高,因此提取溶剂选择50%乙腈溶液。文献[14-15]对诃子化学成分分析均采用70%甲醇溶液作为提取溶剂,笔者在制备诃黎勒酸、诃子酸对照品时发现,两者在甲醇中不稳定,会与溶剂发生可逆的络合反应,因此采用甲醇为溶剂进行提取对含量测定结果影响较大。
2.3 色谱条件优化
2.3.1 检测波长
分别对诃黎勒酸、诃子酸对照品进行200~400 nm 吸收光谱扫描,结果表明,诃黎勒酸最大吸收波长为278 nm,诃子酸最大吸收波长为280 nm,最终检测波长确定为280 nm。
2.3.2 流动相
由于样品提取试剂为乙腈,首先选择乙腈-水作为淋洗液,发现在不同配比条件下1,2,3,4,6-O-五没食子酰基葡萄糖与诃子酸均无法分离,表明1,2,3,4,6-O-五没食子酰基葡萄糖与诃子酸在乙腈中选择性相似,色谱行为相近。在洗脱体系中加入少量甲醇,结果表明,随着甲醇加入量的增加,两者的分离度逐渐增大,当甲醇体积分数为6%时,两者可以达到完全分离,且诃黎勒酸与诃子酸在24 h内稳定性良好,故最终选择以水-乙腈-甲醇三相组成淋洗液。
取诃子药材,按1.3 方法制备样品溶液,对淋洗液组成进行优化,考察两种目标成分的色谱分离效果,两种目标成分保留时间、色谱分离度、理论塔板数及拖尾因子结果见表2。由表2可知,当选择序号为1、3、5的流动相时,诃黎勒酸、诃子酸的分离度较小;选择序号为4的流动相时,诃子酸的保留时间过长;选择序号为7 的流动相时,拖尾因子变大;序号为6 的流动相中增加了甲酸体积分数,但对整体色谱行为影响不大。综合考虑,序号为2 的流动相效果最佳,故流动相选择0.1%的甲酸水溶液-乙腈-甲醇(体积比为78∶16∶6)。
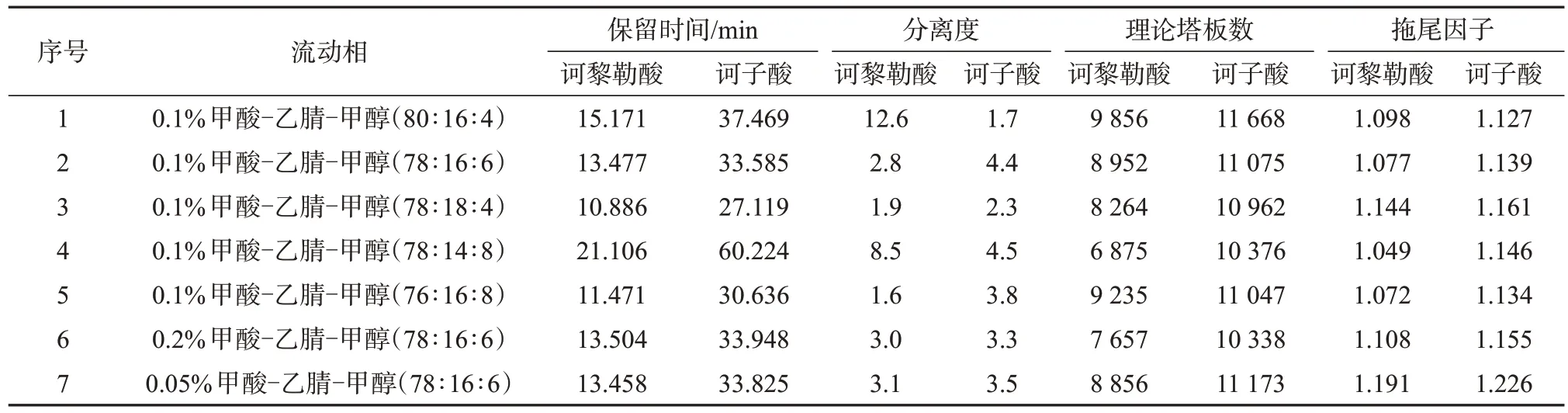
表2 不同流动相组成时诃黎勒酸、诃子酸的色谱分离效果Tab.2 Chromatographic separation effect of chebulagic acid and chebulinic acid under different mobile phase compositions
2.3.3 淋洗液流量
取诃子药材,按1.3方法制备样品溶液,分别选择不同淋洗液流量进行试验,考察两种目标成分的色谱分离效果,两种目标成分保留时间、色谱分离度、理论塔板数及拖尾因子结果见表3。由表3可知,淋洗液流量大于1.0 mL/min时,诃黎勒酸分离度低于2.0;当淋洗液流量小于1.0 mL/min时,诃子酸的保留时间过长。综合考虑,淋洗液流量选择1.0 mL/min。

表3 不同淋洗液流量时诃黎勒酸、诃子酸的色谱分离效果Tab.3 Chromatographic separation effect of chebulagic acid and chebulinic acid at different flow rates of eluent
2.3.4 进样体积
取诃子药材,按1.3 方法制备样品溶液,分别选择不同进样体积进行试验,考察两种目标成分的色谱分离效果,两种目标成分色谱分离度、理论塔板数及拖尾因子结果见表4。由表4可知,当进样体积不大于5 μL 时,诃黎勒酸、诃子酸的分离度均大于1.5;理论塔板数均大于8 000,拖尾因子均小于1.2;但进样体积为3 μL 太小,误差较大;当进样体积为10 μL 时,理论塔板数显著降低;当进样体积为20 μL 时,诃黎勒酸与诃子酸前延峰拖尾现象严重,不能满足定量分析要求。综合考虑,选择进样体积为5 μL。

表4 不同进样体积时诃黎勒酸、诃子酸的色谱分离效果Tab.4 Chromatographic separation effect of chebulagic acid and chebulinic acid at different injection volumes
2.3.5 柱温
取诃子药材,按1.3 方法制备样品溶液,分别选择不同柱温进行试验,考察两种目标成分的色谱分离效果,两种目标成分保留时间、色谱分离度、理论塔板数及拖尾因子结果见表5。由表5可知,在30~40 ℃范围内随着柱温的升高,诃黎勒酸、诃子酸保留时间逐渐减小,其它指标无明显变化,为减少仪器运行的损耗,选择柱温为30 ℃。

表5 不同柱温时诃黎勒酸、诃子酸的色谱分离效果Tab.5 Chromatographic separation effect of chebulagic acid and chebulinic acid at different column temperatures
2.4 系统适用性试验
取对照品溶液、样品溶液分别按1.2色谱条件进样测定,结果如图1所示。由图1可以看出,在样品溶液和对照品溶液的色谱图中诃黎勒酸、诃子酸色谱峰保留时间一致,且实现基线分离。诃黎勒酸保留时间为13.507,分离度为2.2,理论塔板数为8 860,拖尾因子为1.16;诃子酸保留时间为33.711,分离度为3.2,理论塔板数为11 087,拖尾因子为1.20。

图1 诃黎勒酸、诃子酸混合对照品溶液、诃子样品溶液色谱图Fig.1 Chromatogram of mixed reference solutionof chebulagic acid and chebulinic acidand sample solution of Terminalia Chebula Retz.
2.5 线性方程、检出限与定量限
取混合对照品溶液,以50%乙腈溶液逐级稀释成诃黎勒酸质量浓度分别为0.205 0、0.102 5、0.051 3、0.025 6、0.012 8、0.006 4 mg/mL,诃子酸质量 浓 度 分 别 为0.227 5、0.113 8、0.056 9、0.028 4、0.014 2、0.007 1 mg/mL 的系列混合对照品工作溶液。按1.2 色谱条件分别进样测定,以目标物质量(μg)为横坐标,对应色谱峰面积为纵坐标,进行线性回归,计算得诃黎勒酸的线性方程为y=1 591 948x-25 820,相关系数为0.999 6;诃子酸的线性方程为y=2 267 031x-61 935,相关系数为0.999 5。表明诃黎勒酸、诃子酸的质量分别在0.032 03~1.025 μg、0.035 55~1.137 μg 范围内与色谱峰面积线性关系良好。
将质量浓度最低点的混合对照品溶液以50%乙腈溶液逐级稀释,分别进样检测,以信噪比为3∶1时对应的溶液中目标物的质量为方法检出限,以信噪比为10∶1 时对应的溶液中目标物的质量为定量限。诃黎勒酸的检出限为2.67 ng,定量限为8.00 ng;诃子酸的检出限为4.44 ng,定量限为11.85 ng。
2.6 精密度试验
取诃子药材,按1.3 方法平行制备6 份样品溶液,按1.2色谱条件分别进样测定,计算诃黎勒酸和诃子酸的质量分数及相对标准偏差,结果列于表6。由表6可知,诃黎勒酸平均质量分数为97.9 mg/g,相对标准偏差为1.8%;诃子酸平均质量分数为83.9 mg/g,相对标准偏差为1.8%。表明该方法精密度良好。

表6 精密度试验结果Tab.6 Results of precision test
2.7 加标回收试验
取诃子药材样品粉末,按照所建方法测定诃黎勒酸、诃子酸含量,然后取约0.05 g样品,共9份,精密称定,分别置于具塞锥形瓶中,精密加入相当于药材中目标物含量约80%、100%和120%的诃黎勒酸、诃子酸对照品,每个剂量3 份,按1.3 方法制备样品溶液,按1.2色谱条件进样测定,计算两种目标成分的回收率,结果列于表7。由表7可知,诃黎勒酸、诃子酸的回收率均在97%~103%之间,表明方法准确度良好。

表7 加标回收试验结果Tab.7 Results of spiked recovery test
3 结语
建立高效液相色谱法测定中药材诃子中诃黎勒酸和诃子酸的含量,该方法稳定可靠,专属性强,灵敏度高,精密度及准确度好,可对《中国药典》2020年版一部中诃子含量测定项进行补充,以利于诃子药材的质量控制。
