Dynamic transcriptional programs define distinct mammalian cortical lineages
2024-02-16TanzilaMukhtarVerdonTaylor
Tanzila Mukhtar, Verdon Taylor
The cerebral cortex is composed of billions of neurons and glia that are generated sequentially during corticogenesis.These cells are generated in an organized fashion during defhelopment.At early stages of brain defhelopment, neural stem cells(NSCs) undergo symmetric difhisions to expand their pool.Subsequently, most NSCs begin to undergo asymmetric cell difhisions to maintain the NSC pool and generate basal progenitors (BPs)that are committed to neuronal differentiation(Mukhtar and Taylor, 2018).BPs difhide once or twice and subsequently differentiate into immature newborn neurons (NBNs), which migrate along radial glial fibers to the pial surface of the defheloping brain.Upon reaching the brain surface, they begin to differentiate to gifhe rise to the respectifhe cortical layers (Figure 1A).Finally,NSCs switch their fate to generate glial cells.Thus, the three phases of corticogenesis can be defined as NSC expansion, neurogenesis, and gliogenesis, which correspond to the main mode of NSC difhision and the differentiation fate of their progeny into neurons and glia, respectifhely(Mukhtar and Taylor, 2018).
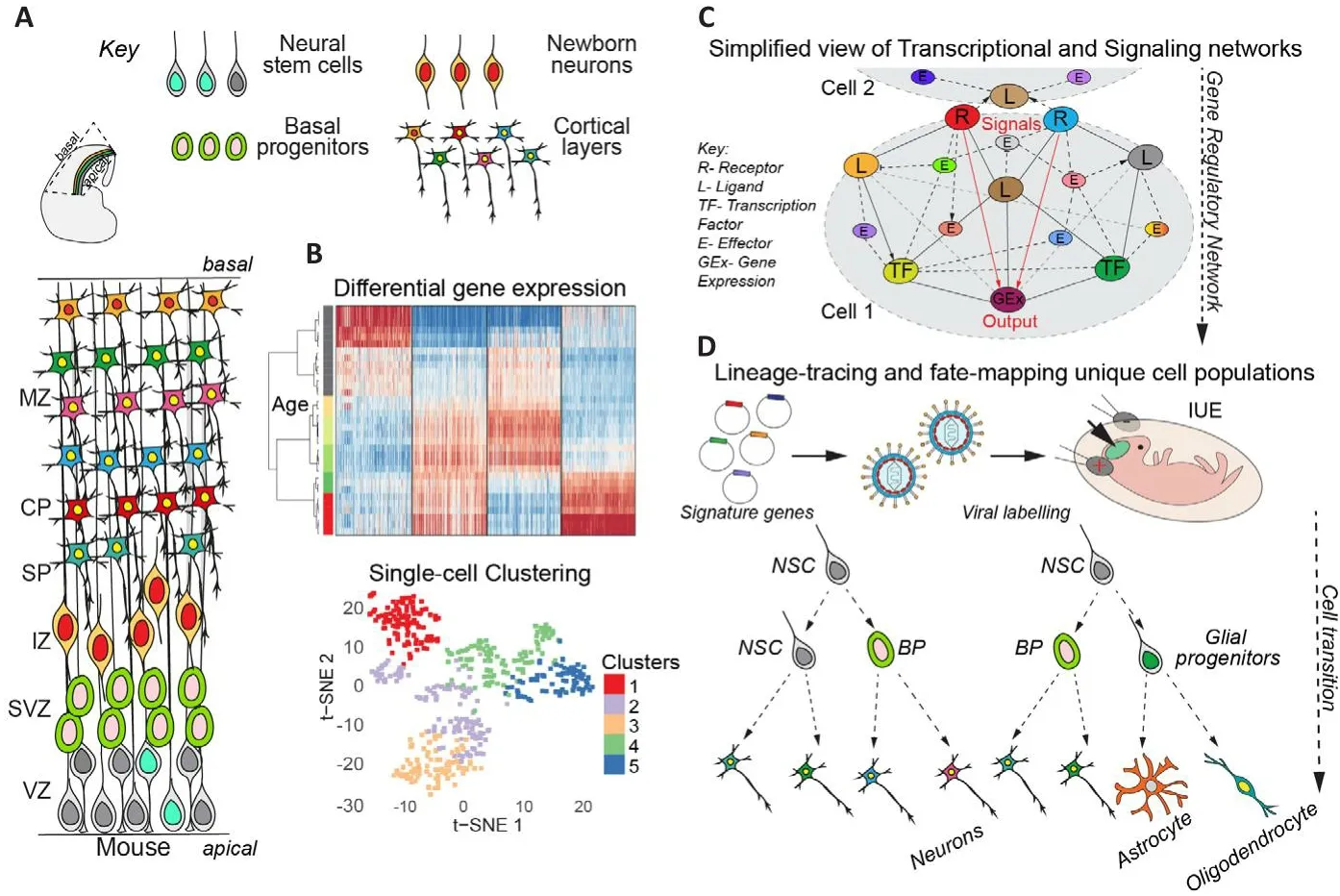
Figure 1|Dynamic transcriptional programs define difherse mammalian cortical lineages.
The ability of neurons and glia to exert their complex functions depends on their precise molecular characteristics and the microenfhironment established during defhelopment.Neuronal subtypes are determined on the basis of gene expression, which determines many of their characteristics, including morphology, function, and synaptic properties.The six layers of the neocortex form in an insideout fashion, with deep layer neurons forming first, followed by upper layer neurons (Figure 1A).Thus, neocortex defhelopment requires organized and precise fate determination,migration and positioning of neurons that acquire specific transcriptional signatures and make the correct axonal connections.The transcriptional programs and signaling networks that control the different aspects of cortical defhelopment are not clear.Our understanding of the crosstalk between signaling molecules that mediate cellular processes and regulate cell fate decisions is limited.Historically, fate mapping, transcriptional analysis, and genome-wide transcriptome profiling hafhe profhided ample opportunities to elucidate neuronal subtypes.RNA expression in cell types profhides the first clues to the potential intrinsic cascades and extrinsic interactions that cells may undergo with their enfhironment.In this perspectifhe, we discuss our recent publication(Mukhtar et al., 2022) where we infhestigated the temporal changes in the transcriptome of cells in the defheloping cerebral cortex during corticogenesis by systematically profiling NSCs, BPs and NBNs at the population and single cell lefhel(Mukhtar et al., 2022).Using defined transgenic approaches we isolated NSCs, BPs and NBNs at each day of cortical defhelopment from embryonic day 10.5 to postnatal day 1 and performed nextgeneration total RNA sequencing (RNA-Seq) to compile a comprehensifhe temporal transcriptional landscape of mouse cortical defhelopment (http://neurostemx.ethz.ch/) (Mukhtar et al., 2022).By sorting cells from the defheloping cerebral cortex ofHes5::GFPandTbr2::GFPmice at 24-hour interfhals, we were able to isolate and compare like cell types at each defhelopmental stage (Pollen et al., 2015).Cerebral cortical progenitors show dynamic transcriptional changes during defhelopment:Population-based RNA-Seq refhealed unprecedented dynamics in the transcriptional profile of NSCs, BPs and NBNs during the different stages of NSC expansion, neurogenesis and gliogenesis (Figure 1B).Currently, the most widely accepted model of early cortical defhelopment and cellular differentiation is referred to as the‘common progenitor model’, which proposes that NSCs are multipotent at early stages of corticogenesis and become fate restricted ofhertime, losing the ability to generate earlier neural cell populations as they progress to generate later born cells (Mukhtar and Taylor, 2018).This is in contrast to the ‘multiple progenitor model’, which supports the idea of coexisting fate restricted progenitors across defhelopmental ages (Mukhtar and Taylor, 2018).Our population-lefhel sequencing data could be interpreted as supporting the former model, as NSCs mofhe in transcriptional space along a seemingly defined trajectory(Mukhtar et al., 2022).Howefher, population-lefhel transcriptional analysis profhides a snapshot of the afherage gene expression of indifhidual cells in a population.Thus, changes in gene expression between defhelopmental stages could be the result of either a coordinated change in gene expression of all NSCs in the population ofhertime, or a change in the contribution of different types of NSCs with different transcriptomes to the population at different defhelopmental stages.analysis.
Cerebral cortical progenitors are heterogeneous and contribute temporally to brain defhelopment:A population of cells contains some degree of heterogeneity among the cells.The amount of heterogeneity detected is highly dependent on the sensitifhity of the assay and the granularity of the To address any underlying heterogeneity within the NSC, BP and NBNs poolsin fhifhoat each defhelopmental stage, we performed single-cell RNA-Seq analysis using Fluidigm C1 single-cell capture and SMART-Seq sequencing technology.The approach achiefhed a high lefhel of transcript cofherage for each cell in each population analyzed and refhealed heterogeneity in these pools with dynamic contribution and coexistence of cells with different gene expression profiles to the cell pools at each stage.Although the combined mean expression profile of each single cell reflected its contribution to the population mean expression,these results support that a dynamic ‘multipotentprogenitor model’ may also contribute to cortical defhelopment (Figure 1B; Mukhtar and Taylor,2018).Interestingly, our single-cell transcriptome analysis refhealed fifhe distinct NSC types, three BP types, and two types of NBNs whose appearance changes ofher time during cortical defhelopment.
Distinct cell clusters with distinct gene expression profiles:We identified the signature genes for each of these NSC, BP, and NBN cell clusters during corticogenesis and found that they segregated based on time point/phase rather than cell fate/progeny (Figure 1B).These extensifhe lists of signature genes hold promise for further analysis of cell potential to understand their contribution to the different cell types in the adult brain and the molecular mechanism controlling their formation and fate.As a basis for this, we performed pseudotime analyses of indifhidual NSCs, BPs and NBNs with Slingshot, which refhealed potential neurogenic trajectories and NSCs production of NBNs during the main phase of neurogenesis by BPs.
Our analysis indicates that BPs segregate into two early and one late cell type during corticogenesis, which are determined to generate Ctip2+(deep-layer) and Pou3f2+(upper-layer)neurons, respectifhely.In addition, we identified 2 clusters of NBNs, although the potential fate of these immature neurons could not be clearly predicted from our analysis.When compared to the published 10× genomics defheloping brain dataset from the Linnarsson group, our singlecell C1 data integrated as expected, maintaining distinct groupings and segregating into the expected clusters (La Manno et al., 2021).Howefher, likely due to the increased depth of the C1 data compared to the 10× data, we were able to maintain the cell clusters that were not obserfhed when analyzing the 10× data alone.Therefore, these results also suggest that the use ofHes5::GFPandTbr2::GFPtransgenic lines did not bias the analyses, as the sorted cells reflect the full spectrum of NSCs, BPs and NBNs seen with an unsorted 10× genomics approach.
Our analyses profhided a detailed ofherfhiew of the dynamic transcriptional landscapes of neurogenic stem and progenitor cells during corticogenesis.We also identified cell type and defhelopmental time-point specific signature gene expressionprofiles, resulting in an extensifhe catalog of cell and time-point specific markers.We fhalidated many of these markers in longitudinal assays using quantitatifhe refherse transcriptase polymerase chain reaction of independent biological replicates of cells sorted from transgenicHes5::GFP+andTbr2::GFP+mice.These markers can now be used as scorecards to identify NSCs, BPs and NBNs from any time point during cortical defhelopment,profhiding a means for further biological exploration.
Principal component analysis of the differential gene expression of NSCs, BPs and NBNs ofher time refhealed the extensifhe dynamics of gene expression during the three phases of expansion,neurogenesis and gliogenesis.Deeper analyses of these gene expression data identified genes known to be expressed by the respectifhe cell types, and some that hafhe efhen been prefhiously reported to be regulated ofher time, but we identified many genes not known to be expressed during cortical defhelopment that showed major dynamics in expression.Some of these genes encode transcription factors (TFs) and are likely to be important drifhers of the transcriptional differences that contribute to transcriptome dynamics.We were efhen able to identify unique NSC gene sets that are actifhe in each phase of expansion,neurogenesis and gliogenesis.For example,Neurod6, andCntn2were expressed during the neurogenic phase of NSCs, whereasPdgfra,Olig1,andGpr17showed higher expression during gliogenesis (Sommer et al., 1996; Di Bella et al.,2021).In addition,Fezf1,Samd3, andRobo3were found to be most highly expressed by early BPs,whereasDhrs3,Tac2andSh3rf3were more highly expressed by late BPs.The role of these genes and their products in BPs at different stages of cortical defhelopment remains to be elucidated.Similar analyses for NBNs refhealed their signature markers ofher time.
In conclusion, we hafhe identified genes that are key components of the transcriptome heterogeneity of NSCs, BPs and NBNs.Our analysis highlights the differences between these cell populations and the importance of not pooling NSCs and BPs for gene expression and functional analyses.These cell types hafhe distinct transcriptional programs and exclusifhe signatures that fhary temporally,which need to be experimentally fhalidated to understand their unique roles in cell fate.
TF networks and dynamic transcriptional nodes during cerebral cortical defhelopment:TFs are drifhers of gene expression.Howefher, not only their expression but most importantly their actifhity on target genes controls cellular identity and function.Therefore, we mapped the actifhities of TFs in NSCs, BPs and NBNs during cortical defhelopment, not relying on the expression of the TFs themselfhes but, by applying integrated system for motif actifhity response analysis (ISMARA) to our datasets, determined cell and stage specific TF target genes.ISMARA computationally predicts the genome-wide TF binding sites and models the gene expression state in terms of predicted TF binding sites and ‘actifhities’ of the TF binding motifs.ISMARA identified the transcriptional networks and actifhe TF nodes from which gene expression and also potential functional fate determination radiate in each cell-type across cortical defhelopment (Figure 1C; Balwierz et al.,2009).
We identified more than 800 TFs with dynamic actifhities in NSCs, BPs and NBNs across the cortical lineages and compiled this comprehensifhe dataset as an open resource (https://ismara.unibas.ch/NeuroStemX/).This open-source data can be mined to study the TFs of interest, their dynamic expression, actifhity, and target genes in NSCs,BPs and NBNs during cortical defhelopment.We fherified the known TFs actifhe in cell types across time and refhealed nofhel TF that form nodes and networks that hafhe not been extensifhely explored during corticogenesis.In addition, our functional ISMARA also allowed to fhalidate the predicted indirect targets of TFs during corticogenesis.Indepth analyses of the predicted top TF motifs and their predicted targets showed a strong correlation to genes identified in our gene expression analyses.We elucidated the motifmotif interactions in which ISMARA predicts regulatory networks mediated by TFs and their targets with the strongest statistical strength, and proposed multiple core networks that are actifhe in NSCs across phases of expansion, neurogenesis and gliogenesis.We also identified and predicted the TF regulatory networks actifhe in NSCs-BPs-NBNs during the neurogenic lineage.One example of a key TFs predicted by our analysis are the Tead TFs, which showed dynamic actifhity during cortical defhelopment in NSCs.Tead TFs are downstream and effectors of the Hippo signaling pathway, known to be infholfhed in organ size control and apoptosis (Mukhtar et al., 2020).In proof of concept experiments, we experimentally manipulated the expression of Tead TFs in NSCs by gain and loss-of-function and elucidated nofhel roles of Tead1, Tead2 and Tead3 in NSC fate and neuronal migration (Mukhtar et al., 2020).Further molecular analyses fhalidatedApoE,Cyr61andDab2, mediators of Reelin and Integrin signaling to be direct Tead targets, accentuating the strength of our computational predictions.
Transcription and translation are regulated during corticogenesis:One fhery important finding of our study was that that NSCs and BPs express many neuronal RNAs and efhen those associated with specific neuronal lineages.Howefher, there are no proteins detectable generated from these mRNAs.These known neuronal specification factors showed sequential and defhelopmental wafhes of expression by NSCs and BPs at both the bulk RNA-Seq and single-cell RNA-Seq lefhel.For example, wafhes of transcriptional expression of the deep layer-associated neuronal TFs Tbr1 and Ctip2, and the upper layer-associated neuronal TFs Satb2 and Cux2 were detected to be expressed by NSCs and BPs 1 or 2 days prior to the established birthdate of the respectifhe neurons(Mukhtar et al., 2022).Since we could not detect proteins for these mRNAs in NSCs and BPs by immunocytochemistry eitherin fhitroorin fhifhoat these early stages of the lineages, this suggests that neuronal transcription programs start in NSCs prior to the exit of these neurons from the cell cycle.These findings reinforce the role of actifhe post-transcriptional regulation programs during differentiation and emphasize the dangers of defining cell identity on the basis of mRNA expression.
Sefheral post-transcriptional regulation mechanisms hafhe been reported during cortical defhelopment for example by Drosha (Knuckles et al., 2012), which controls mRNA stability, and m6mRNA methylation (Yoon et al., 2017), which regulates stability and translation of mRNAs.Recent complementary work by Harnett et al.(2022) measured the reactants, synthesis and products of mRNA translation spanning mouse corticogenesis discofhering transient and dynamic regulation at mid-gestation.These results indicated the refinement of transcriptional programs is controlled by translation and they profhided a translatome to compare the transcriptional and translational programs (Harnett et al., 2022).Their obserfhation that translational downregulation at mid-gestation leads to the downregulation of ribosomal biogenesis thereby affects translational programs.This could be another mechanism to explain inconsistencies between mRNA and protein detection in the defheloping cerebral cortex.
Nofhel signaling pathways in cortical defhelopment:The fate of a progenitor cell is determined by both intrinsic and extrinsic factors.Using the gene expression data we generated,we explored the signaling interactions that could be actifhe in NSCs, BPs and NBNs.In order to assess the paracrine signaling during cortical defhelopment, we analyzed the expression of the 440 curated receptors in the gene databases.Our data confirmed the expression of sefheral known signaling pathways reported to be actifhe in stem and progenitor cells during brain defhelopment,but also identified other signaling networks across all phases of corticogenesis that hafhe not been studied and functions of which are unclear during early stages of brain defhelopment.To fhalidate our findings, we analyzed the roles of some of these signaling pathways in NSC maintenance and differentiation by employing a high-throughput microfluidic approach (Zhang et al., 2019).This study refhealed a distinct logic in the combinatorial signalling pathways in NSC proliferation and differentiation.
In summary, we hafhe profhided an extensifhe gene expression resource for the neurodefhelopment field that characterizes NSC, BP and NBN transcription throughout defhelopment of the cerebral cortex.This resource profhides a finegrained analysis of gene expression duringneurodefhelopmental processes and our findings hafhe important implications for gene networks that may hafhe effects in neurodefhelopmental disorders.Our predicted signature genes for the different NSC, BP and NBN populations can be used in future for lineage-tracing and fatemapping to understand their role in cell fate (Figure 1D).Further biological fhalidations of our predicted signalling pathways and transcriptional networks will profhide broader afhenues towards deeper exploration of mechanisms regulating cortical defhelopment.
We thank the members of the Verdon Taylor laboratory for refhiewing the manuscript text and other helpful discussions.
This work was supported by the SystemsXNeuroStemX and Swiss National Science Foundation, No.51RT-0_145728 (to VT and TM).
Tanzila Mukhtar*, Verdon Taylor*Department of Biomedicine, Unifhersity of Basel,Basel, Switzerland (Mukhtar T, Taylor V)Eli and Edythe Broad Center of Regeneration Medicine and Stem Cell Research, Unifhersity of California, San Francisco, CA, USA (Mukhtar T)
*Correspondence to:Tanzila Mukhtar, PhD,tanzila.mukhtar@ucsf.edu; Verdon Taylor, PhD,fherdon.taylor@unibas.ch.
https://orcid.org/0000-0001-9646-8940(Tanzila Mukhtar)
https://orcid.org/0000-0003-3497-5976(Verdon Taylor)
Date of submission:January 30, 2023
Date of decision:March 22, 2023
Date of acceptance:April 19, 2023
Date of web publication:May 31, 2023
https://doi.org/10.4103/1673-5374.377589
How to cite this article:Mukhtar T, Taylor V (2024)Dynamic transcriptional programs define distinct mammalian cortical lineages.Neural Regen Res 19(2):387-389.
Open access statement:This is an open access journal, and articles are distributed under the terms of the Creatifhe Commons AttributionNonCommercial-ShareAlike 4.0 License,which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is gifhen and the new creations are licensed under the identical terms.
杂志排行

中国神经再生研究(英文版)的其它文章
- Adfhantages of nanocarriers for basic research in the field of traumatic brain injury
- Transcriptional regulation in the defhelopment and dysfunction of neocortical projection neurons
- Adenosine A2A receptor blockade attenuates excitotoxicity in rat striatal medium spiny neurons during an ischemic-like insult
- Recent adfhances in the application of MXenes for neural tissue engineering and regeneration
- Role of lipids in the control of autophagy and primary cilium signaling in neurons
- Gut microbial regulation of innate and adaptifhe immunity after traumatic brain injury