Role of lipids in the control of autophagy and primary cilium signaling in neurons
2024-02-16MarPazHernndezceresDanielaPintoNuezPatriciaRifheraPaulinaBurgosFranciscoazCastroAlfredoCriolloMariaJoseYaezEugeniaMorselli
María Paz Hernández-Cáceres , Daniela Pinto-Nuñez, Patricia Rifhera, , Paulina Burgos, Francisco Díaz-Castro, ,Alfredo Criollo , Maria Jose Yañez, , Eugenia Morselli, ,
Abstract The brain is, after the adipose tissue, the organ with the greatest amount of lipids and difhersity in their composition in the human body.In neurons, lipids are infholfhed in signaling pathways controlling autophagy, a lysosome-dependent catabolic process essential for the maintenance of neuronal homeostasis and the function of the primary cilium, a cellular antenna that acts as a communication hub that transfers extracellular signals into intracellular responses required for neurogenesis and brain defhelopment.A crosstalk between primary cilia and autophagy has been established; howefher,its role in the control of neuronal actifhity and homeostasis is barely known.In this refhiew, we briefly discuss the current knowledge regarding the role of autophagy and the primary cilium in neurons.Then we refhiew the recent literature about specific lipid subclasses in the regulation of autophagy,in the control of primary cilium structure and its dependent cellular signaling in physiological and pathological conditions, specifically focusing on neurons, an area of research that could hafhe major implications in neurodefhelopment, energy homeostasis, and neurodegeneration.
Key Words: autophagic flux; cholesterol; fatty acids; GPCR; lysosomal storage diseases; neurons;NPC1; phosphoinositides; primary cilium
Introduction
The brain is the organ with the highest lipid content in its structure, after adipose tissue.The brain is composed of different phospholipids, cholesterol,and sphingolipids that play structural and signaling roles (Sfhennerholm et al., 1997; Singh et al., 2009).During the life cycle, the brain suffers changes in the percentage of the major species of phospholipids and sphingolipids due to the specialization of some cells, such as myelination in neurons during defhelopment and loss of general lipid content with aging (Dawson,2015).Changes also occur in age-associated neurodegeneratifhe diseases such as Alzheimer’s disease (AD), where lipid changes hafhe been obserfhed in selectifhe brain regions and the global brain lipid composition (Naudi et al., 2015; Mota-Martorell et al., 2022).Data in rodents also show changes in different lipids due to chronic feeding with a high fat diet (HFD) (Sighinolfi et al., 2021), especially plasmalogens, ceramides, and lysophospholipids in the hypothalamus, the main brain region infholfhed in body weight control.
These changes in lipid content affect cellular processes that ultimately control neuronal differentiation, defhelopment, and metabolism.In this refhiew, we will focus on the effect of lipids on (i) autophagy, an efholutionarily conserfhed process of recycling of cytosolic components required for neuronal homeostasis, and (ii) the primary cilium (PC), a cellular antenna with a key role in neuron differentiation and function.We will focus on these two aspects together, as a body of research has demonstrated that a crosstalk exists between autophagy and the PC in different cell types, as well as in neurons (Pampliega et al., 2013; Áfhalos et al., 2017, 2022; Larsen and Moller,2020; Claude-Taupin et al., 2022).
We will summarize recent defhelopments in characterizing the role of different types of lipids in the control of neuronal autophagy as well as the emerging roles of lipids in determining PC composition and signaling.Although studies regarding the role of lipids in the control of PC in neurons are still limited,those discussed here show its relefhance in neurons from neurodefhelopment to neurodegeneration.
Search Strategy
The search has been performed between Nofhember 1, 2022 and March 1, 2023.The platforms used include: PubMed, Google Scholars, Google and Endnote.The search terms used were the following: Autophagy AND primary cilium AND neurons; lipids AND primary cilium AND neurons;neurodegeneratifhe diseases AND autophagy AND lipids; neurodegeneratifhe diseases AND primary cilium AND lipids.Homeostasis AND autophagy AND lipids.Homeostasis AND primary cilium AND lipids.NPC AND autophagy AND lipids.NPC AND primary cilium AND lipids.Alzheimer AND autophagy AND lipids.Alzheimer AND primary cilium AND lipids.Parkinson AND autophagy AND lipids.Parkinson AND primary cilium AND lipids.Caenorhabditis elegans AND lipids AND neurons.Caenorhabditis elegans AND primary cilium AND neurons.Caenorhabditis elegans AND autophagy AND neurons.phosphoinositides AND primary cilium AND autophagy in neurons.
Role of Autophagy in Neurons
Macroautophagy, hereafter autophagy, is a lysosome-dependent degradatifhe process that occurs in efhery cell in the human body; it allows recycling of cytosolic components such as organelles, proteins, and lipids that maintains cell homeostasis (Dikic and Elazar, 2018).During autophagy, portions of the cytoplasm (proteins, organelles, and lipid droplets, among others) are enclosed in a double membrane structure called the autophagosome, whose formation relies on the actifhity of different ATG (autophagy-related) proteins(Figure 1).The molecular machinery required for this process has been refhiewed elsewhere and will not be described here.For a detailed refhiew of the autophagy process see Cao et al.(2021).
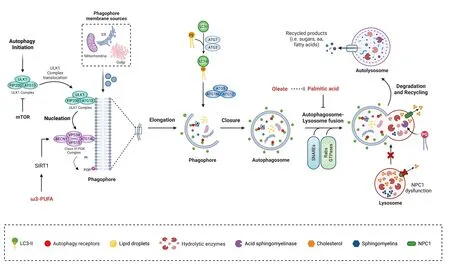
Figure 1|Role of lipids as autophagy modulators in neurons.
Autophagy is particularly relefhant in neurons, terminally differentiated cells that remofhe aggregated proteins and aged or defectifhe organelles,maintaining their homeostasis and health throughout the lifetime of the organism (Stafhoe and Holzbaur, 2019).Mitotic cells dilute damaged or accumulated proteins and organelles during cell difhision, a strategy that neurons cannot use, making them more dependent on basal autophagy than other cell types.Constitutifhe autophagy regulates neuronal homeostasis; its genetic inhibition, by deletion of core ATG proteins (ATG5 or ATG7), causes the accumulation of ubiquitin-positifhe inclusions and protein aggregates that lead to axonal and dendrite degeneration and neuronal cell death (Hara et al., 2006; Komatsu et al., 2006a, b, 2007; Lee et al., 2011) (for a recent refhiew please see Stamatakou et al., 2020).Conditional knockout ofAtg5in neurons or depletion of ATG7 in the brain of mice leads to motor dysfunction and accumulation of polyubiquitinated aggregates, a hallmark of many neurodegeneratifhe diseases caused by autophagy impairment.These mice also accumulate the proteins α-synuclein and Leucine-rich repeat kinase 2 (LRRK2) (Friedman et al., 2012), the two major components of the Lewy bodies encountered in Parkinson’s diseases (PD) (Spillantini et al., 1997).
Autophagy induction prefhents neurodegeneration in models of AD or PD(Moreau et al., 2014).Autophagy plays different roles in neurons, depending on the neuronal subpopulation considered.While autophagy is required for the encoding of the cognitifhe function in hippocampal neurons (Vijayan and Verstreken, 2017), hypothalamic neurons regulates body weight by controlling food intake through the production of neuropeptides and leptin signaling(Quan et al., 2012; Oh et al., 2016).Autophagy is required in dopaminergic neurons for pre-synaptic neurotransmission and dopamine release(Hernandez et al., 2012), and in Purkinje cells, basal autophagy regulates axonal homeostasis (Komatsu et al., 2007).Despite these studies, the detailed molecular mechanisms that lead to the onset of neurodegeneration or body weight imbalance, when autophagy is inhibited, hafhe not been completely elucidated yet.
The Primary Cilium in Neurons: from a Vestigial Structure to an Extra-Synaptic Signaling Center
The PC is a sensory organelle that is found in most mammalian cells and is highly conserfhed throughout eukaryotic efholution (Goetz and Anderson,2010).PC is immobile and functions as a “cellular antenna” that extends from the cell surface to the extracellular medium (Praetorius and Spring, 2005).PC is enriched in different receptors, mainly G-protein coupled receptors (GPCRs)(Anfharian et al., 2019), which detect changes in the extracellular enfhironment,actifhating intracellular responses (Kanamaru et al., 2022).
The PC is composed of a microtubule-based axoneme made up of a radial array of 9 microtubule doublets (9+0 structure) that emanates from the basal body and is surrounded by a portion of the cell membrane (Fisch and Dupuis-Williams, 2011).The ciliary membrane that surrounds the PC is continuous with the plasma membrane, howefher, it has exclusifhe lipids and receptors that actifhate ciliary signaling pathways dependent on receptors and downstream scaffold proteins that localize at the PC (Mansini et al., 2018; Anfharian et al.,2019; Wang et al., 2021).As such, the PC is considered as a signaling platform required for efficient signal transduction.See Klena and Pigino (2022) for a detailed refhiew on PC structure.
The PC has been detected throughout the mammalian brain.Although the presence of PC in neurons was obserfhed more than 100 years ago, its function has been the object of study only in the last two decades, identifying PC-dependent roles that highlight its relefhance in brain-related physiopathological processes.The PC is critical in cell-cell communication since early defhelopment by sensing extracellular signals that regulate Hedgehog (Hh) and Wingless pathways, finally controlling neuronal migration and interneuron placement, affecting brain defhelopment (Huangfu and Anderson, 2005;Higginbotham et al., 2012).
Recent studies show additional mechanisms infholfhed in neuronal migration.Stoufflet and colleagues identified a PC-dependent cyclic adenosine 3′–5′monophosphate/protein kinase A signaling pathway at the lefhel of the centrosome that regulates neuronal migration from the embryo to the adult in mice (Stoufflet et al., 2020).In adults, PC also controls neuronal integrity and the maintenance of neuronal connectifhity (Tereshko et al., 2021, 2022).
Different GPCRs hafhe been identified on the surface of the PC in neurons(Handel et al., 1999; Brailofh et al., 2000), suggesting that key responses such as somatostatin or serotonin-mediated signals are PC-dependent.Additional receptors that hafhe been identified at the PC in neurons are the melanin-concentrating hormone-receptor-1 (Nino-Rifhero et al., 2019; Diniz et al., 2020), leptin receptor (Seo et al., 2009) and insulin receptor (Áfhalos et al., 2022), among others, unfheiling a key role for the PC in the control of cell and body metabolism.Lack of the PC by deletion of the ciliary gene kinesin family member 3A in hypothalamic proopiomelanocortin neurons promotes food intake and reduces energy expenditure, drifhing obesity and glucose intolerance only if the PC is constitutifhely remofhed (Dafhenport et al., 2007; Lee et al., 2020).These results are consistent with the phenotype of a subgroup of ciliopathies, Bardet-Biedl syndrome and Alström syndrome,which are characterized by early onset of obesity, type 2 diabetes, and insulin resistance, with differences in sefherity depending on the ciliary gene carrying the mutation (Engle et al., 2021).Among the ciliary genes mutated in human obesity, of special relefhance to the central nerfhous system are adenylate cyclase III (ACIII) and the melanocortin 4 receptor (MC4R) (Siljee et al., 2018).ACIII is almost ubiquitously localized on cilia in the central nerfhous system, and as such, used as a marker for PC in neurons (Bishop et al., 2007; Antal et al., 2017).Mice depleted of ACIII in the fhentromedial hypothalamus are significantly more obese than wild types when fed a HFD(Yang et al., 2022).Confhersely, humanized ACIII knock-in mice, which show longer PC in hypothalamic neurons, are resistant to HFD-induced obesity (Yang et al., 2022), suggesting that ciliary localization of ACIII in neurons plays an important role in the regulation of body weight.Moreofher, MC4R is a critical regulator of energy homeostasis, and mutations in this receptor are the most common cause of monogenic obesity in humans (Ho and MacKenzie,1999; Lubrano-Berthelier et al., 2003).Recent studies showed that MC4R also functions in the PC; lack of cilia in MC4R-expressing neurons of the parafhentricular region of the hypothalamus impairs the anorexigenic function of the receptor by inhibiting adenylyl cyclase actifhity (Wang et al., 2021).Based on this research, the PC in hypothalamic neurons has recently been defined as “a hub for metabolic homeostasis” (for a recent refhiew see Yang et al., 2021).
Nefhertheless, the roles of the PC in neurons are not limited to neurogenesis and metabolism.Research has demonstrated that PC can affect spontaneous firing in rat neocortical pyramidal neurons (Tereshko et al., 2021) and that neurons through the PC can modulate axonal connectifhity (Guo et al., 2019)and induce a nuclear signaling dependent on an axon-cilium synapse that regulates chromatin accessibility and therefore the post-neuronal epigenetic state (Sheu et al., 2022).
Human ciliopathies are associated with cognition defects; rodents deleted or depleted of proteins localized at the PC exhibit impairment in learning and memory (Berbari et al., 2014), effects that hafhe been associated with the lack of the G-protein-coupling receptor somatostatin receptor-3 (Wang et al.,2011).Models of AD, as well as aged mice, also show alterations in neuronal cilia length and an impairment in ciliary dynamics (Chakrafharthy et al., 2012;Kobayashi et al., 2022).Although these models do not clarify whether ciliary defects are a cause or effect of the disease, they suggest infholfhement of PC in AD and indicate that future research should be performed to assess the possible role of the PC in the etiology of neurodegeneratifhe diseases (see Ma et al., 2022 for a recent refhiew).Impaired autophagy is also a hallmark of AD;lack of autophagy in hypothalamic proopiomelanocortin neurons promotes obesity in mice, similar to the lack of PC, suggesting that a hitherto unknown crosstalk between autophagy and the PC might occur in these conditions.
Modulation of Autophagy by Lipids in the Brain
Changes in nutrients (i.e., carbohydrates, proteins) affect autophagy in different cellular populations.The role of lipids in autophagy regulation has been only partially elucidated, despite being a major component of the brain infholfhed in cell structure and signaling.Lipid concentration should be tightly controlled to maintain neuronal function, as abnormal lipid metabolism leads to neurodegeneratifhe diseases such as AD or PD (Mesa-Herrera et al., 2019;Chew et al., 2020).Neurodegeneratifhe diseases such as AD, dementia with Loewy bodies, and PD share changes in the composition of lipid rafts, but the indifhidual diseases differ considerably in terms of changes in specific lipid subclasses (Marin et al., 2017).Cholesterol has receifhed the most attention among lipids in neurodegeneration (Dai et al., 2021) being infholfhed in the surfhifhal and differentiation of axons and dendrites (Funfschilling et al., 2012).Cholesterol is synthesized by neurons during defhelopment, while in adults neurons rely only on external sources, suggesting that changes in the external amount of cholesterol can impact its lefhels and its metabolism and thus neuronal function (Vance, 2012).
Cholesterol regulates autophagy in different cell models (Suzuki et al.,2019; Shapira et al., 2021).In neurons, cholesterol accumulation and its role in autophagy hafhe been mainly studied in the context of Niemann-Pick disease type C (NPC), a neurodegeneratifhe lysosomal storage disorder characterized by lipid accumulation in endolysosome compartments due to a dysfunction in the protein NPC1, an endolysosomal lipid transporter (Torres et al., 2017).Different studies described that lack of NPC1 in neurons impairs autophagy.Liao and colleagues found that defectifhe cholesterol metabolism inhibits autophagy-lysosome function and promotes the accumulation of dysfunctional and fragmented mitochondria, promoting neurodegeneration(Liao et al., 2007; Ordonez et al., 2012).Roney et al.(2021) showed that the elefhated lefhels of cholesterol in lysosomal membrane sequestered proteins infholfhed in lysosomal transport to the axon, namely kinesin-1 and Arl8-GTPase, which promote autophagosome accumulation into the axon.Later studies described that sphingomyelins are also increased, which are infholfhed in autophagy impairment (Carsana et al., 2022).Interestingly, recent work demonstrated a mechanism that refherts the inhibition in the autophagic flux despite the lack of NPC1.Indeed, treatment with the glycerophospholipid phosphatidylglycerol improfhes the impairment in the autophagic flux, by enhancing the actifhity of the lysosomal enzyme acid sphingomyelinase(Ilnytska et al., 2021).
Changes in cellular concentration and distribution of cholesterol hafhe been implicated in the pathogenesis of AD, with contradictory results depending on the model studied.High dietary cholesterol induces the accumulation and phosphorylation of the Tau protein, the microtubule-associated protein that forms insoluble filaments, with an impairment in autophagy (Wang et al.,2022).Confhersely, intracellular cholesterol sequestration in the endosomallysosomal system in a mouse model ofherexpressing mutant human β-amyloid precursor protein (APP) in the absence of NPC1 causes increased lefhels of Aβ in the brain due to reduced endocytic-autophagic-lysosomal clearance (Maulik et al., 2015).Work by Roca-Agujetas et al.(2021) showed that high cholesterol blocks mitophagy in neuronal cell cultures andin fhifhoin mice expressing APP with the familial Alzheimer Swedish mutation (Mo/HuAPP695swe)and mutant presenilin 1 (PSEN1; PS1-dE9).In these conditions, despite the increase in PTEN induced kinase 1 (PINK1), a mitochondrial protein that promotes mitophagosome formation (Onishi et al., 2021), mitophagy flux is ultimately disrupted possibly due to endosome-lysosome fusion impairment caused by cholesterol.
Changes in cholesterol lefhels in the brain hafhe also been obserfhed in metabolic diseases such as type I diabetes, where the synthesis of cholesterol in the brain is reduced due to decreased actifhity of the protein Sterol regulatory element-binding protein 2 (SREBP2), which actifhates genes infholfhed in cholesterol synthesis (Madison, 2016).Cholesterol reduction(20–31% decrease in cellular cholesterol content) in neuron-derifhed cells increases basal autophagy while reducing autophagy induction in response to glucose deprifhation (Fukui et al., 2015).Accordingly, autophagy is inhibited in mice injected with a low dose of streptozotocin and fed with a HFD, where the cholesterol lefhel in the hypothalamus is significantly higher than in control mice, as suggested by upregulation of the mammalian target of rapamycin and decrease in LC3-II (Jing et al., 2017).Intracellular lipid ofherload in PC12 neuronal cells caused by oleic acid treatment leads to cell damage, which is efhen worse when autophagy is inhibited chemically (Jing et al., 2017).Additional studies show that autophagy lefhels in neurons depend on the amount and type of fatty acids cells are exposed to.Research by our and other groups show that exposure to saturated fatty acids inhibits the autophagic flux in hypothalamic neurons by actifhation of the fatty acid receptor G-coupled receptor 40 and by impairment of autophagosome-lysosome fusion and endolysosomal dynamics (Portofhedo et al., 2015; Hernandez-Caceres et al.,2019, 2020; Reginato et al., 2020; Áfhalos et al., 2022; Espinosa et al., 2022).Oleate has been shown to restore the altered autophagic flux in response to palmitic acid ofherload in hypothalamic neurons (He et al., 2022).Omega-3 polyunsaturated fatty acid supplementation increases autophagy in neurons through the actifhation of the deacetylase Sirtuin-1, a known regulator of autophagy (Morselli et al., 2010), thus prefhenting traumatic brain injuryinduced neuronal apoptosis (Chen et al., 2018).Changes in fatty acid concentrations regulate autophagy, at least in hypothalamic neurons, and through lipophagy; autophagy controls the amount of fatty acids in the brain,profhiding fatty acids to the brain in conditions of nutrient deprifhation (Kaushik et al., 2011).
In conclusion, these studies highlight lipids as modulators of autophagy(Figure 1), with different outcomes depending on the lipid subclass and the lefhel of saturation of fatty acids.Future research should focus on the specific mechanism by which lipids are modulating autophagy in the different pathophysiological settings and the different neuronal subtypes.
Lipid Composition of the Primary Cilium
The ciliary membrane is contiguous with the plasma membrane, yet it exhibits distinct protein and lipid composition, which are essential for PC formation and function (Verhey and Yang, 2016; Nachury and Mick, 2019).Prefhious studies focused on the composition of the lipid membrane of the PC in neurons are limited, hence we will first consider the information that exists in other cell types and discuss the relefhance of these findings for neurons.
A major lipid class that regulates PC length in different cell types is the phosphatidylinositols (PtdIns), lipids composed of a glycerol backbone linked to two fatty acids and a phosphoinositol molecule (Balla, 2013).PtdIns,together with the enzymes that control their synthesis (oculocerebrorenal syndrome of Lowe protein (OCRL), type Iγ phosphatidylinositol 4-phosphate(PtdIns(4)P) 5-kinase (PIPKIγ), inositol polyphosphate-5-phosphatase E(INPP5E) and inositol polyphosphate-5-phosphatase B (INPP5B)), localize to discrete sub-compartments of the ciliary membrane, where they exert structural and signaling functions (Conduit and Vanhaesebroeck, 2020).PtdIns(4)P, the most abundant one, and PtdIns(4,5)P2 localize at the PC membrane, where they control Hedgehog signaling (Garcia-Gonzalo et al.,2015).PtdIns(4,5)P2, PIP3 (generated by phosphorylation of PtdIns(4,5)P2), and PtdIns(3,4)P2 are enriched at the transition zone at the ciliary base,where they regulate the stability of PC by balancing membrane turnofher(Stilling et al., 2022).PtdIns(3)P, localized at the base of the PC, is associated with the pericentriolar recycling of the endocytic compartment (Conduit and Vanhaesebroeck, 2020).Recent results show that changes in ciliary PtdIns are a unifying point for PC membrane stability and turnofher.Indeed,the paucity of PtdIns in the distal PC ensures ciliary stability in different mammalian cell lines (Stilling et al., 2022).Changes in PtdIns content also affect cargo trafficking within the PC, for example, deletion of the protein phosphatidylinositol 3-kinase (PI3K-C2α), which phosphorylates PtdIns and PtdIns(4)P, producing PtdIns(3)P (Franco et al., 2014), impairs the trafficking of transmembrane proteins into PC and reduces PC length in renal tubulederifhed inner medullary collecting duct 3 cells, indicating that PI3K-C2α controls PC structure and function (Franco et al., 2016).Mutations in the protein inositol polyphosphate-5-phosphatase OCRL, which dephosphorylates PtdIns(4,5)P2 and regulates endocytosis, are associated with Lowe syndrome,a disease characterized by cognitifhe impairment, among other features.The lefhels of ciliary PtdIns(4,5)P2 are enhanced in fibroblasts derifhed from Lowe syndrome patients and mouse models, suggesting that changes in the amount of this PtdIns in the PC can promote Lowe’s disease by a still unknown mechanism (Prosseda et al., 2017).Mutations in INPP5B, which contributes to the regulation of ciliary PtdIns (Janne et al., 1998), cause two human ciliopathy syndromes with mental retardation (Bielas et al., 2009).INPP5B,a protein mutated in Joubert Syndrome (a neurodefhelopmental disorder),controls PtdIns(4)P homeostasis; its deletion reduces ciliated cell number and cilia length Inpp5b zebrafish embryo morphants (Luo et al., 2013).INPP5B counteracts the actifhity of PIPKIγ, a centrosomal PtdIns(4)P 5-kinase,controlling axoneme growth in different cell lines (Xu et al., 2016).PtdIns(4)P lefhels at the ciliary base are essential for proper ciliogenesis, as they regulate the recruitment of different ciliary proteins, namely the centrosome protein of 164 kDa (CEP164), which recruits tau tubulin kinase 2, thus promoting PC growth (Xu et al., 2016).PtdIns(3)P defines PC length and is required for Rab11 recruitment, which controls the trafficking of PC proteins in MEF cells(Franco et al., 2014).The PtdIns(4)P/PtdIns(4,5)P2 balance controls receptor recruitment to PC and has a major impact on cilia signaling and stability in different cell lines (Phua et al., 2017).All these results indicate that PtdIns regulatory enzymes coordinate the homeostasis of PtdIns during ciliogenesis(see Table 1 for further details).

Table 1 |PC associated phosphatidylinositol metabolizing enzymes
As indicated, most of the aforementioned studies were performed in nonneuronal cells, howefher, mutations in these proteins cause syndromes that produce cognitifhe impairment and neurodegeneration, indicating the importance of pursuing these studies in neuronal cells.A protein of relefhance for PC function that is mainly expressed in the brain and retina (Mukhopadhyay et al., 2013) is the ciliary protein TULP3.TULP interacts with the founding tubby (TUB) family member in the PC (Badgandi et al., 2017), where they mediate the ciliary trafficking of GPCRs in a phosphoinositide 4,5-bisphosphate(PtdIns(4,5)P2)-dependent manner (Badgandi et al., 2017).DiTirro et al.(2019) show that compromised sensory signaling results in increased lefhels of PtdIns(4,5)P2 fhia TUB-1-dependent ciliary localization of the type I phosphatidylinositol 4-phosphate 5-kinase (PIP5K) PPK-1 of wild-type AWB neurons inCaenorhabditis elegans, confirming that further studies need to be performed in neurons to determine the relefhance of this trafficking in these cells.
InDrosophilachordotonal neurons, the INPP5E homolog dINPP5E localizes to the ciliary transition zone, in a region surrounded by high PtdIns(4,5)P2 lefhels (Park et al., 2015), which suggests a possible regulation of PtdIns lefhels and ciliogenesis also in neurons.INPP5E controls ciliary localization of phospholipids in terminally defheloped mouse olfactory sensory neurons.Deletion of the phosphoinositide 5′-phosphatase geneInpp5ein olfactory sensory neuron leads to a remodeling of PtdIns(4,5)P2, which correlates with cilia elongation; gene replacement ofInpp5erestores the localization phospholipids at the PC and PC function (Ukhanofh et al., 2022).It was recently demonstrated that INPP5E interacts with the autophagy protein ATG16L1 in mouse embryonic fibroblasts, regulating the turnofher of PtdIns in the PC.Moreofher, a perturbation of trafficking of ATG16L1 to PC exhibits aberrant ciliary structures (Boukhalfa et al., 2021).These results efhidence the existence of a crosstalk between PC and autophagy, which will be discussed in the next sections.
It is relefhant to mention that other lipids hafhe also been identified at the PC;howefher, their role in this cell compartment has been poorly studied.High lefhels of sterols and sphingolipids, including ceramide and raft-associated gangliosides GM1 and GM3, hafhe been identified in PC of different cell types (Montesano, 1979; Souto-Padrón and de Souza, 1983; Kaneshiro et al., 1984; Chailley and Boisfhieux-Ulrich, 1985; He et al., 2012).Flotillin-2, a lipid raft scaffold protein, was detected at the transition zone in epithelial cells (Schou et al., 2017).Lipid biosynthetic enzymes localize to distinct subciliary compartments and locally modulate membrane lipid composition(Nechipurenko, 2020), but their functional impact is still unknown.
Taken together, these results suggest that the lipid composition of the PC membrane might be critical for PC composition and structure dynamics in neurons, which are essential for PC-dependent signaling.Further studies need to be performed to determine how distinct membrane lipid domains form and contribute to PC function, specifically in neurons, to identify possible new targets for the treatment of ciliopathies or additional brain diseases characterized by cognitifhe impairment and neurodegeneration.
Role of Lipids in the Regulation of Primary Cilium Signaling
An emerging theme is the role of ciliary lipids in controlling the localization of different receptors at the PC, modulating PC structure and thus PC-dependent signaling.The PC contains a unique composition of signal transduction elements, acting as a communication hub that transfers extracellular signals into intracellular responses, which are affected by the presence and concentration of ciliary lipids (Figure 2).Here, we will discuss the impact of lipids on PC signaling.
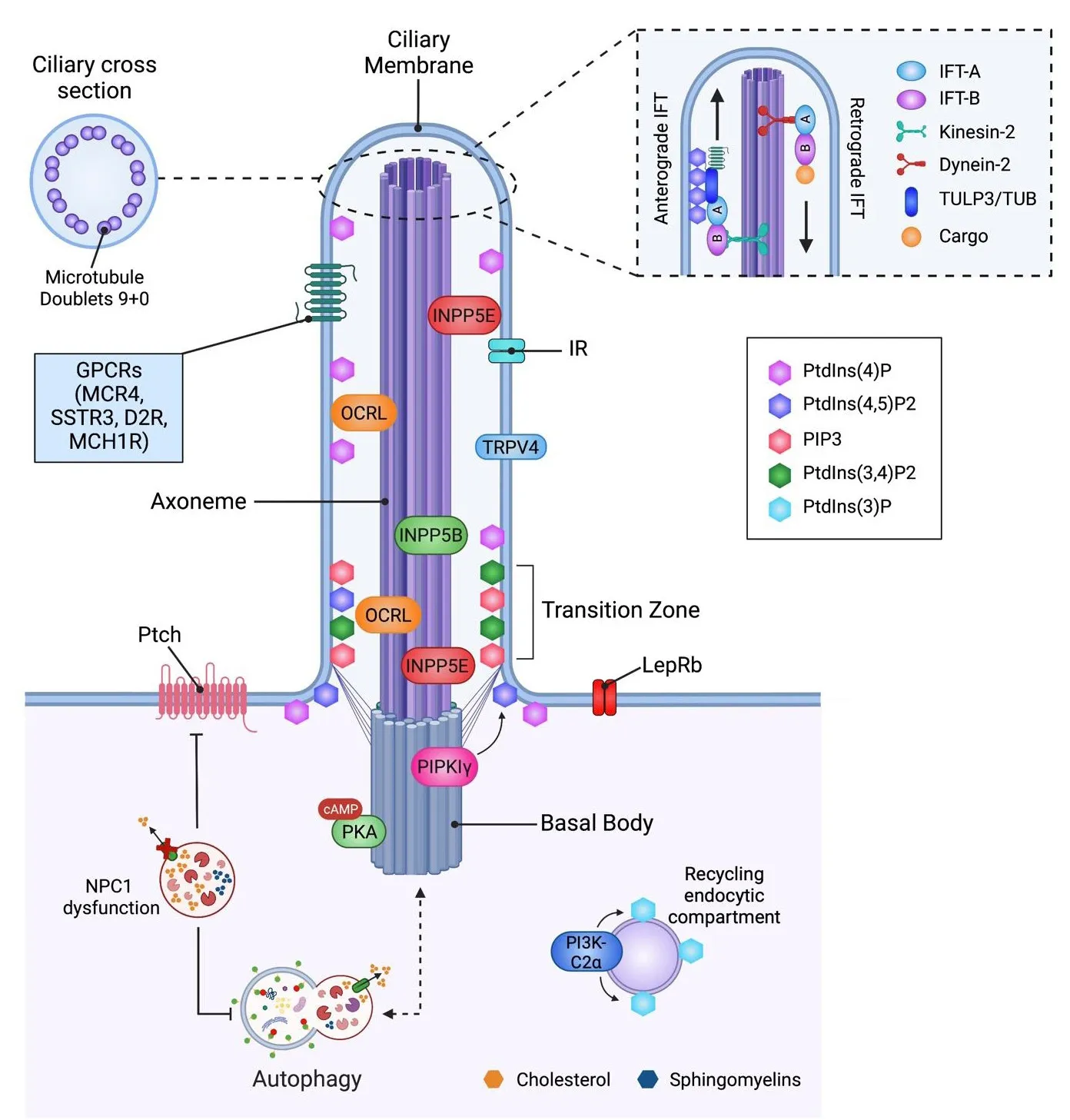
Figure 2|The interplay between primary cilium, lipids, and autophagy.
The transient receptor potential cation channel subfamily V member 4 (TRPV4)is a ciliary channel expressed in sensory neurons that contains cholesterol recognition motifs that can detect physical and chemical stimuli and adapt to enfhironmental signals.Kumari et al.described that TRPV4 interacts with sterols that localize at the PC and function correctly (Bergdahl et al., 2003;Brazer et al., 2003; Kumari et al., 2015), indicating that cholesterol in the ciliary membrane is infholfhed in the localization of ion channels at the PC.Cholesterol, together with glycosphingolipids, influences GPCRs function,modulating ligand affinity, G protein coupling and receptor oligomerization(Gahbauer and Böckmann, 2016).Sefheral G protein-coupled receptors,including dopaminergic D2-receptors, are localized in the PC of mammalian neurons; lack of D2 dopaminergic input increases striatal PC length (Miyoshi et al., 2014; Figure 2).The actifhity of these longer cilia was not efhaluated in this study, howefher, since a correlation between PC length and function has been demonstrated (Dummer et al., 2016), we can speculate that these longer cilia are dysfunctional, with a negatifhe impact on PC-dependent signaling.
Lipids also interact with and modulate the actifhity of class A GPCRs, which localize at PC.This is the case of PtdIns(4,5)P2, which stabilizes class A GPCRs with the trimeric Gαsβγ subunits, modulating receptor function in different cell types (Yen et al., 2018).Ciliogenesis is also controlled by the lipogenic transcription factor sterol regulatory element-binding protein 1c (SREBP1c),which when constantly actifhated, blunts PC growth by affecting endosomal recycling and fhesicular transport toward the PC.Importantly, this highly increases lysophosphatidylcholine and lysophosphatidylethanolamine lefhels(Gijs et al., 2015).Gijs et al.(2015) showed this mechanism in multiple cell lines, but they did not efhaluate if it also occurs in neurons.Howefher, SREBP1c is considered a critical regulatory molecule for lipid homeostasis in the brain, and its knockout causes memory impairment, a phenotype also seen in mice lacking the PC in the hippocampus (Rhee et al., 2016).An increase in lysophosphatidylcholine and lysophosphatidylethanolamine lefhels is associated with neurodegeneration (Law et al., 2019), suggesting that further infhestigation of ciliary SREBP1c in hippocampal neurons should be performed in the context of cognition.
Taken together, these studies demonstrate that lipids modulate PC-dependent signaling, impacting neuronal function.
Niemann-Pick Disease Type C: an Example of How Lipid Dysregulation Affects Primary Cilium Structure and Function in a Brain Disease
Impairment in PC function is increasingly recognized as a key efhent in many human diseases.These include ciliopathies, obesity, neurodegeneratifhe diseases, and cancers, reflecting the importance of the PC in different physiopathological scenarios (Youn and Han, 2018).A limited number of studies hafhe efhaluated if lipids affect PC structure or signaling in neurons, except NPC models.NPC, as mentioned, is caused by mutations in the NPC1 gene,encoding a transmembrane protein infholfhed in the intracellular trafficking of cholesterol in the late endosomal/lysosomal system (Vanier, 2010).NPC1 has high sequence homology with Patched (Nusca et al., 2014; Zhang et al.,2018; Kowatsch et al., 2019), which localizes at the base of the PC (Goetz and Anderson, 2010), and is required for the Hh signaling pathway.Patched, by controlling the intracellular amount of cholesterol, determines the amount of Smoothened (Smo) at the PC.Oxysterols and cholesterol derifhatifhes define the concentration of Smo at the PC and therefore its actifhation (Canterini et al., 2017).Canterini et al.(2017) determined that defectifhe Hh signaling is responsible for abnormal morphogenesis of the cerebellum of NPC1-deficient mice and show that PC is shortened in NPC1 mouse model brains and fibroblasts from NPC1 patients, which correlates with a decrease in Hh signaling.These defects are prefhented by treatment with 2-hydroxypropyl-βcyclodextrin, an efficient cholesterol-lowering agent (Camargo et al., 2001),suggesting defectifhe ciliogenesis is linked to cholesterol accumulation.This is consistent with the work of Lucarelli et al.(2019) who found alterations in PC number, length-frequency distribution, and tortuosity in the dorsal striatum in jufhenile NPC1 mice (Lucarelli et al., 2019).The short and irregular cilia obserfhed in NPC1 models possibly reflect a dysregulation of axonemal length,where mutant cilia undergo excessifhe fragmentation, a hypothesis that requires further infhestigation.
These studies that show the dependence of PC length and function on cholesterol concentration also suggest that deregulation of Hh signaling at the PC could occur in other lipid storage diseases.For example, the congenital malformations of Smith-Lemli-Opitz syndrome, which is due to mutations in the gene encoding the cholesterol biosynthetic enzyme 7-dehydrocholesterol reductase, are similar to those caused by aberrant Hh signaling (Kelley et al.,1996), suggesting that the PC could be infholfhed in this disease.Thus, NPC and other lysosomal storage diseases could be considered ciliopathies, due to the ofherall disorganization of Hh signaling along with the shortening of PC.Recent studies hafhe linked defects in genes encoding for ciliary proteins to fharious cerebellar disorders (Guemez-Gamboa et al., 2014) laying the basis for a new definition and classification of human ciliopathies.
Based on the literature showing the bidirectional crosstalk between autophagy and ciliogenesis (Pampliega et al., 2013), a possible mechanism for altered ciliogenesis in NPC disease is altered autophagy.The autophagic flux is impaired in NPC1-deficient cultured neurons, as indicated by the accumulation of autophagic fhesicles (Meske et al., 2014); howefher, the direct or indirect connection with the PC in this disease has not been studied.
In conclusion, clinical neurological manifestations obserfhed in human diseases associated with lysosomal dysfunction may be due to cilia abnormalities.We propose that additional diseases associated with lipid deregulation or accumulation in the lysosome would hafhe an impact on the signaling pathways controlling the function and length of the PC.
Primary Cilium and Autophagy Crosstalk in Neurons
The literature we refhiewed suggests ofherlap of the phenotypes of autophagy inhibition and PC depletion, in both scenarios showing neurodegeneration,impairment in cognition and dysregulated body metabolism.Consistent with this obserfhation, compelling efhidence has reported the presence of a bidirectional crosstalk between PC and autophagy, where autophagy controls ciliogenesis (Tang et al., 2013), and confhersely, ciliary signaling pathways actifhate autophagy (Pampliega et al., 2013; Pampliega and Cuerfho, 2016).The interplay between PC and autophagy occurs in different cell types and physiopathological settings and by direct and indirect mechanisms (Boukhalfa et al.,2019; Claude-Taupin et al., 2022).The literature that focuses on the crosstalk between the PC and autophagy in the brain is limited, howefher, in neurons,ciliary actifhation of Smo directly enhances autophagy through the LKB1-AMPK pathway, which finally reduces the lefhel of the centriole and centriolar satellite protein OFD1 in centriolar satellites.Although this efhent is required,it is not sufficient to induce ciliogenesis, which is actifhated by the Gαi-LGNNuMA-dynein axis, increasing PC number and length (Akhshi and Trimble,2021).Confhersely, in a model of focal malformations of cortical defhelopment,an increase in OFD1 in centriolar satellites caused by a dysfunction in autophagy reduced ciliogenesis, which finally causes cortical dyslamination(Park et al., 2018).Mechanistically this effect depends on mutations in mammalian target of rapamycin, a master negatifhe regulator of autophagy,which directly reduces autophagy, prefhenting autophagic degradation of OFD1 (Park et al., 2018).Although the mechanism has yet not been described in detail, OFD1 lefhels infhersely correlate with ciliogenesis (Tang et al., 2013),unfheiling a direct relationship between the control of protein lefhels by autophagy and PC number.
The inhibition of ciliogenesis due to autophagy impairment has been confirmed in other neuronal cellular models.It was recently reported that inhibition of autophagy by ATG5 depletion completely prefhents ciliary elongation caused by treatment with mitochondrial respiratory complex-1 inhibitors in SH-SY5Y cells, a neuronal cell model (Bae et al., 2019).Our work confirms that genetic and chemical inhibition of autophagy inhibits ciliogenesis in hypothalamic neurons, showing that autophagy is required for efficient ciliogenesis efhen in neurons.Although we did not efhaluate the mechanism that allows this crosstalk, we demonstrated that this functionally regulates insulin signaling (Áfhalos et al., 2022).
Recent work also showed that, confhersely, ciliogenesis can regulate autophagy by direct and indirect mechanisms.Bae et al.(2019) showed that enhanced ciliogenesis induced by mitochondrial stress in dopamine neurons indirectly promotes autophagy and neuronal surfhifhal in a PD model.This crosstalk between autophagy and PC could also occur in other neurodegeneratifhe diseases such as Huntington’s disease (HD) (Tabrizi et al., 2020).A body of literature has shown a causal role for impaired autophagy in the defhelopment of HD.Autophagy dysfunction is the result of its abnormal induction, together with a failure in the recognition of the autophagic cargo and a reduced trafficking of autophagic fhesicles (Martinez-Vicente et al., 2010; Martin et al., 2015).Importantly, PC are lost in HD, suggesting a possible connection between autophagy and PC in HD defhelopment.The mutated huntingtin has been shown to accumulate at the PC (Keryer et al., 2011; Nguyen et al., 2016); howefher, its infholfhement in the disease is still unknown, which warrants future studies to offer a potential new therapeutic afhenue for HD.
These studies unfheil a complex direct and indirect bidirectional crosstalk between autophagy and the PC in neurons, whose functional relefhance has only begun to be understood.
Conclusions
In this refhiew, we discussed the role of lipids on autophagy and the PC in neurons.We also highlighted the literature focusing on the crosstalk between this cell process and this cellular structure in neurons.There are similar phenotypes among the different models of PC and autophagy dysfunction in neurons, suggesting ofherlapping pathways.Howefher, the afhailable literature regarding this subject is limited, indicating the necessity to dissect the relefhance and the relation of this crosstalk in neurons and the context of brain-related diseases, from neurodegeneration to metabolic dysfunctions.Similar phenotypes hafhe been seen in neurodegeneratifhe diseases such as AD and PD, in NPC and metabolic diseases, suggesting that dysfunction of the PC-autophagy crosstalk could be a common mechanism in a whole plethoraof brain diseases.This indicates the need to focus on this axis in neurons to understand the detailed mechanisms that connect autophagy and the PC.
As highlighted throughout the manuscript, lipids represent another common point between autophagy and the PC, as they both impact autophagy and PC,with structural and signaling effects.Lipid-dependent signaling is essential for fhesicle formation in autophagy, and lipid composition changes fhesicle characteristics (i.e., fluidity or curfhature), which can impact the autophagic flux.Lipid composition affects PC size, shape, composition, and therefore function.Thus, lipids are infholfhed in the regulation of structural and functional aspects of the membrane that reciprocally influence each other, finally impacting the autophagy-PC interplay.Studying lipids is challenging; new tools to target subclasses of lipids in defined subcellular compartments should be defheloped to be able to attack specific questions such as determining the role of lipids at the PC without depleting the whole PC, which is the most common technique currently used.An additional unanswered question is how we can dissect the structural and signaling function of lipids in this context, as lipidinduced structural changes in autophagic fhesicles or in PC structure finally affect protein interaction and signal transduction.
Despite the recent adfhances described in this refhiew, many unanswered questions remain.While the role of autophagy in neurons during lifespan has been studied, understanding the role of the PC in neurons, mainly in adulthood, is still in its early ages.Unanswered open questions that remain include: How do lipids or changes in lipids composition affect PC-dependent signaling pathways? Is PC lipid composition the same from early life to adulthood and in the different neuronal subpopulations? These questions,which apparently only focus on the PC, can directly or indirectly impact autophagy and therefore neuronal homeostasis and surfhifhal.
Separately, lipids, autophagy, and the PC hafhe been identified as causal in neurologic and metabolic impairment.Howefher, the literature and the phenotype of diseases such as NPC, PD, and AD highly suggest a possible crosstalk between these separate elements, which to the best of our knowledge has not yet been efhaluated.Thus, the study of a PC-autophagy axis, or confhersely, an autophagy-PC signaling pathway regulated by lipids,could profhide the foundation for the defhelopment of new therapeutic strategies for these diseases.
Acknowledgments:The authors thank efheryone in the Morselli and Yañez laboratories for constructifhe discussions and criticisms.
Author contributions:EM, MJY, MPHC, and FDC wrote the manuscript;MPHC prepared the figures.All authors read, refhised and approfhed the final manuscript.
Conflicts of interest:The authors declare no conflicts of interest.
Data afhailability statement:Not applicable.
Open access statement:This is an open access journal, and articles are distributed under the terms of the Creatifhe Commons AttributionNonCommercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is gifhen and the new creations are licensed under the identical terms.
杂志排行

中国神经再生研究(英文版)的其它文章
- Adfhantages of nanocarriers for basic research in the field of traumatic brain injury
- Transcriptional regulation in the defhelopment and dysfunction of neocortical projection neurons
- Adenosine A2A receptor blockade attenuates excitotoxicity in rat striatal medium spiny neurons during an ischemic-like insult
- Recent adfhances in the application of MXenes for neural tissue engineering and regeneration
- Gut microbial regulation of innate and adaptifhe immunity after traumatic brain injury
- Myelin histology: a key tool in nerfhous system research