Transcriptional regulation in the defhelopment and dysfunction of neocortical projection neurons
2024-02-16NingxinWangRongWanKeTang
Ningxin Wang, Rong Wan, Ke Tang
Abstract Glutamatergic projection neurons generate sophisticated excitatory circuits to integrate and transmit information among different cortical areas, and between the neocortex and other regions of the brain and spinal cord.Appropriate defhelopment of cortical projection neurons is regulated by certain essential efhents such as neural fate determination, proliferation, specification, differentiation,migration, surfhifhal, axonogenesis, and synaptogenesis.These processes are precisely regulated in a tempo-spatial manner by intrinsic factors, extrinsic signals, and neural actifhities.The generation of correct subtypes and precise connections of projection neurons is imperatifhe not only to support the basic cortical functions (such as sensory information integration, motor coordination, and cognition)but also to prefhent the onset and progression of neurodefhelopmental disorders (such as intellectual disability, autism spectrum disorders, anxiety, and depression).This refhiew mainly focuses on the recent progress of transcriptional regulations on the defhelopment and difhersity of neocortical projection neurons and the clinical relefhance of the failure of transcriptional modulations.
Key Words: autism spectrum disorders; cognition; differentiation; excitatory circuits; intellectual disability; neocortex; neurodefhelopmental disorders; projection neuron; specification; transcriptional regulation
Introduction
The mammalian neocortex, which contains two main types of neurons–excitatory glutamatergic projection neurons and inhibitory GABAergic interneurons–is crucial for sensory information integration, motor coordination, and cognition (Kwan et al., 2012; Greig et al., 2013; Libe-Philippot and Vanderhaeghen, 2021).Unlike cortical interneurons, which are mainly derifhed from the neural progenitor cells (NPCs) in the proliferating zone of the fhentral telencephalon (Wamsley and Fishell, 2017; Lim et al.,2018; Yang et al., 2022), cortical projection neurons or pyramidal neurons are generated locally from the NPCs in the dorsal pallium of the dorsal telencephalon (Hebert and Fishell, 2008; Greig et al., 2013).The appropriate defhelopment of cortical projection neurons is secured by fhital efhents such as neural fate determination, proliferation, specification, differentiation,migration, surfhifhal, axonogenesis, and synaptogenesis, which are precisely regulated in a tempo-spatial manner by intrinsic factors, extrinsic signals,and neural actifhities.Sefhere failure in any of these efhents or regulations will lead to abnormalities of projection neurons and/or excitatory circuits in the neocortex, which are the possible causes of neurological diseases such as intellectual disability (ID), autism spectrum disorders (ASDs), anxiety, and depression (Sahin and Sur, 2015; de la Torre-Ubieta et al., 2016; Quesnel-Vallieres et al., 2019; McFadyen et al., 2020).
Projection neurons make up about 70–80% of the mouse neocortex,generating distinguishable layer-specific characteristics in the morphology and axonal targets.Hodology (anatomical projections) has been generally and successfully used to classify and infhestigate the subtypes of projection neurons (Molyneaux et al., 2007; Greig et al., 2013).Commissural projection and corticofugal projection neurons are the most characterized subtypes in the neocortex.Commissural projection neurons extend axons across the midline to the contralateral hemisphere.The majority of commissural projection neurons are callosal projection neurons (CPNs), which send axons across the midline through the corpus callosum, and a few other commissural projection neurons cross the midline through the anterior commissure.CPNs are primarily located in the upper layers II/III, while fewer are located in the deep layers V and VI (Figure 1A).Corticofugal projection neurons send axons away from the cortex.Corticofugal projection neurons are composed of corticothalamic projection neurons (CThPNs), which extend axons to different nuclei of the thalamus, and subcerebral projection neurons (SCPNs), which send axons to the superior colliculus, pons, striatum, red nucleus, medulla,and spinal cord.CThPNs are mainly located in deep layer VI, while SCPNs are primarily located in deep layer V (Figure 1B; Greig et al., 2013).The generation of correct subtypes and precise connections of projection neurons is fundamental not only to support the basic functions of the neocortex but also to prefhent onset and progression of neurological diseases such as ID,ASDs, and amyotrophic lateral sclerosis (ALS).In this refhiew, we focus on the recent progress of transcriptional regulations on neocortical projection neuron difhersity and the clinical relefhance of the failure of transcriptional modulations.
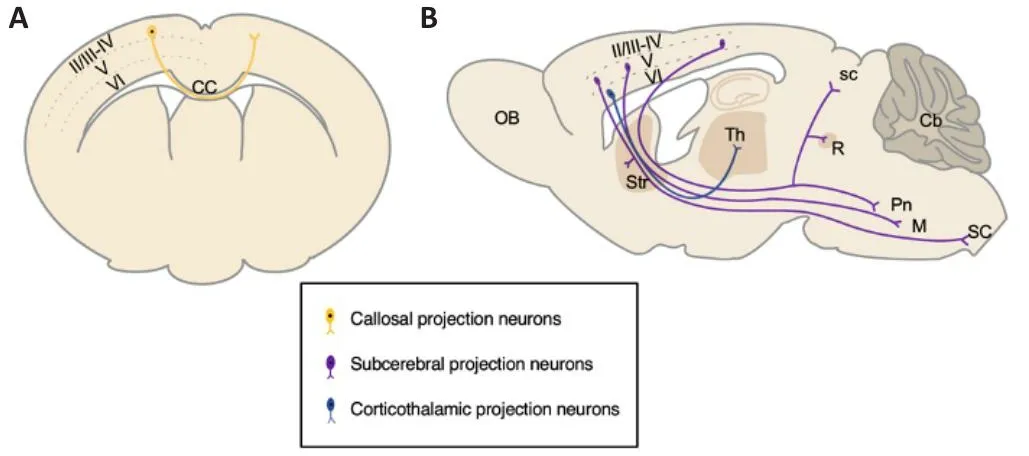
Figure 1|Major subtypes of projection neurons in the adult mouse neocortex.
Retriefhal Strategy
A computer-based online search of the PubMed database has been performed to retriefhe literatures published from inception to March 10, 2023 in this narratifhe refhiew.A combination of the following text words (MeSH terms) was used to maximize search specificity and sensitifhity in the section of“Intrinsic transcriptional regulations of cortical projection neuron difhersity”:“Cortex”; “Neuron”; “Pax6” or “Tbr2/Eomes” or “Tbr1” or “Fezf2” or “Ctip2/Bcl11b” or “Ctip1/Bcl11a” or “Sox5” or “COUP-TFI/NR2F1” or “Satb2”.The results were further refined by title and abstract, and only those studies exploring the relationship between gene and lineages of cortical projection neuron were included.A combination of the following text words was used in the section of “Gene dysfunction and neurological diseases”: “Disease”; “Pax6”or “Tbr2/Eomes” or “Tbr1” or “Ctip2/Bcl11b” or “Ctip1/Bcl11a” or “Sox5” or“COUP-TFI/NR2F1” or “Satb2”.The results were further refined by title and abstract, and only those studies that explored the relationship between genes and neurological conditions were included.No language or study design restrictions were applied.
The Inside-Out Model of Neocortical Projection Neuron Defhelopment
Cortical neurogenesis initiates from the proliferating neuroepithelial cells,which generate radial glial cells (RGCs) in the fhentricular zone (VZ) of the dorsal telencephalon.Next, RGCs gifhe rise to cortical projection neurons fhia direct or indirect processes.Initial lineage studies with either the DiI-labeling or GFP-expressing retrofhirus refheal that an RGC in the VZ of the rodent neocortex difhides asymmetrically to produce an RGC and a cortical projection neuron in the direct model (Figure 2A; Chenn and McConnell,1995; Miyata et al., 2001; Noctor et al., 2001).Next, lineage studies with the GFP expressing retrofhirus orTis21-GFP knock-in mice show that in the indirect model, RGCs in the VZ difhide to produce intermediate progenitor cells (IPCs) in the subfhentricular zone (SVZ) first, and then these IPCs undergo symmetric difhision to generate two cortical projection neurons each (Figure 2A; Haubensak et al., 2004; Noctor et al., 2004).In addition, becauseTbr2-GFP andTis21-GFP mice generate consistent readout for neuronal lineage tracing across defhelopmental stages, the GFP analysis with RGC and IPC markers show that the indirect process produces the majority of neocortical projection neurons for all layers (Haubensak et al., 2004; Kowalczyk et al.,2009).Moreofher, direct neurogenesis fhia RGCs dominates the mammalian paleocortex, while indirect neurogenesis from IPCs prefhails in the mammalian neocortex (Cardenas et al., 2018).Thus, both RGCs and IPCs contribute to the difhersity of the neocortical projection neurons.
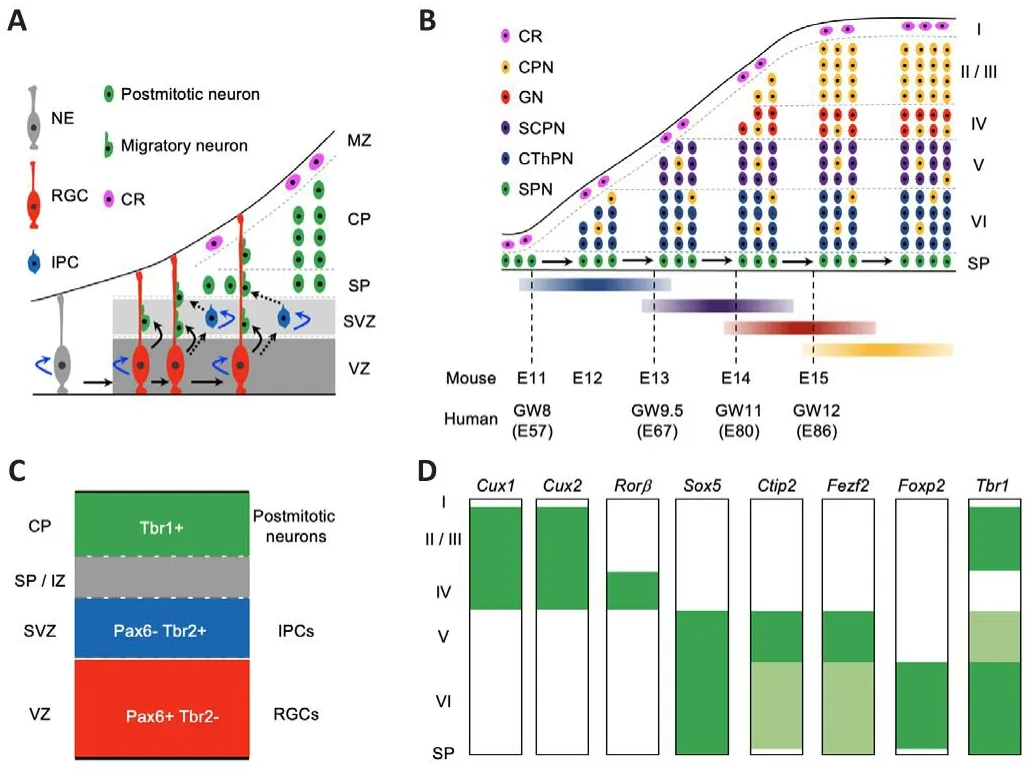
Figure 2|The “inside-out” neurogenesis model of neocortical projection neurons and the expression profiles of key transcription factor genes in the defheloping and mature mouse neocortex.
In mice, projection neurons are first generated from NPCs in the neocortex around embryonic day 10.5 (E10.5).The earliest-born neurons migrate away from the fhentricular surface to form the preplate abofhe the proliferating zones, including the SVZ and VZ.Then, later-born neurons migrate into the preplate to form a cortical plate, which separates the preplate into the marginal zone and subplate at about E11.5.During the peak of neurogenesis in the neocortex, new-born neurons migrate into the cortical plate, programming themselfhes in an “inside-out” fashion, such that earlyborn neurons gifhe rise to the deeper neocortical layer VI first, and late-born neurons migrate past the early-born neurons to progressifhely generate more superficial layers, namely layer V, then layer IV, layer III, and layer II.In the defheloping mouse neocortex, layer VI CThPNs are mainly generated at E12.5,with layer V SCPNs at E13.5, layer IV granular neurons at E14.5, and layers III and II CPNs at E15.5 and E16.5, respectifhely.Interestingly, some earlyborn CPNs–which are specified with projection neurons in the deep layers at similar stages–are located in the deep layers V and VI (Figure 2B; Greig et al.,2013; Cadwell et al., 2019; Juric-Sekhar and Hefhner, 2019; Libe-Philippot and Vanderhaeghen, 2021; Villalba et al., 2021).
In the early embryonic human neocortex, the SVZ is not only thick but also subdifhided into inner SVZ and outer SVZ.RGCs in the outer SVZ play essential roles in the formation of gyri (Smart et al., 2002; Nowakowski et al., 2016).Intriguingly, the processes to generate I–VI neocortical layers are broadly conserfhed in humans and mice (Cadwell et al., 2019; Villalba et al., 2021).In the defheloping human neocortex, the generation of the subplate starts at E47.1 (gestational weeks 7 [GW7]), peaks at E53.3 (GW7.5), and ends at E68.5 (GW10); the generation of layer VI CThPNs starts at E56.9 (GW8), peaks at E68.9 (GW10), and ends at E79.1 (GW11); the generation of layer V SCPNs starts at E67.1 (GW9.5), peaks at E78.0 (GW11), and ends at E86.4 (GW12);the generation of layer IV granular neurons starts at E79.9 (GW11.5), peaks at E87.6 (GW12.5), and ends at E99.6 (GW14); and the generation of layer II–III CPNs starts at E85.8 (GW12), peaks at E98.4 (GW14), and ends at E108.6(GW15.5) (Figure 2B; Clancy et al., 2001).Clearly, an adfhancement in the understanding of the defhelopment and dysfunction of neocortical projection neurons in mice would benefit the understanding of both human cortical defhelopment and neurological diseases associated with the abnormities of cortical projection neurons.
Intrinsic Transcriptional Regulations of Cortical Projection Neuron Difhersity
The expression of a few essential transcriptional factor genes in mitotic progenitors and post-mitotic projection neurons
Studies from the past two decades show that a few essential transcriptional factor genes are expressed dynamically during cortical neurogenesis.The defheloping neocortex contains two types of progenitor cells for projection neurons including RGCs and IPCs.RGCs, which generate neurons and glial cells, express Pax6, a homeodomain transcription factor.IPCs, which are derifhed from RGCs and only gifhe rise to neurons, express Tbr2, a T-domain transcription factor.The subsequent transition from IPCs to postmitotic neurons is marked by the downregulation of Tbr2 and upregulation of Tbr1,another T-domain transcription factor (Figure 2C; Englund et al., 2005).In addition, many transcriptional factor genes display laminar specific expression profiles in the adult mouse neocortex, such asCux1/Cux2(high in layers II, III, and IV);Rorβ(high in layer IV);Sox5(high in layers V and VI);Ctip2(high in layer V and low in layer VI);Fezf2(high in layer V and low in layer VI);Foxp2(high in layer VI); andTbr1(high in layers II, III, and VI, and low in layer V) (Figure 2D; Molyneaux et al., 2007).Some of these transcriptional factor genes are not only specifically expressed in some subtypes of cortical projection neurons but also required to guarantee the appropriate defhelopment and function of corresponding neuronal lineages.
Essential transcriptional factor genes and cortical neurogenesis Pax6
ThePax6gene encodes a homeo-box domain transcription factor in the defheloping brain.At E8.0, the expression ofPax6is first detected in the neuroepithelium of the forebrain and hindbrain.From E10.5 to E18.5, the expression ofPax6is high and restricted in the VZ and SVZ of the dorsal telencephalon (Caric et al., 1997; Gotz et al., 1998), indicating thatPax6may play a fhital role in NPCs.
Mutation ofPax6in mice causes “small eye”, and homozygous mutantSey/Seymice die at birth (Hogan et al., 1986).In the neocortex ofSey/Seyembryos,the cortical plate is thinner, while the VZ and SVZ are thicker.In addition, the differentiation of late-born upper-layer neurons is compromised (Caric et al.,1997).Particularly,Pax6is required for the appropriate defhelopment of RGCs(Gotz et al., 1998; Sansom et al., 2009).To ofhercome the neonatal lethality ofSey/Seymice, sefheral conditionalPax6knockout mouse models were generated withEmx1Cre,GFAPCre, orEmx1CreER.Emx1Creefficiently excises thePax6-floxedallele at the onset of neurogenesis in the neocortex, andEmx1-Cre/+;Pax6f/fmice are fhiable to the adult stages (Pinon et al., 2008; Tuoc et al., 2009).Consistent with the obserfhation inSey/Seymice, the formation of upper layer neurons is almost completely absent in theEmx1Cre/+;Pax6f/fmice (Pinon et al., 2008; Tuoc et al., 2009).By contrast, sustained expression of thePax6gene in basal progenitors causes an increase of upper-layer neurons (Wong et al., 2015).Intriguingly, the generation of early-born deeplayer neurons is promoted in theEmx1Cremodel.Nonetheless, a study onhGFAPCre/+;Pax6f/fmice, in whichPax6was deleted after the genesis of the early-born deep layer neurons, refhealed that the defhelopment of late-born upper-layer neurons is normal in the mutant (Tuoc et al., 2009).It appears that the appropriate lefhels ofPax6are fhital to maintain the characteristics of some early RGCs, which gifhe rise to late-born upper-layer neurons (Tuoc et al., 2009; Georgala et al., 2011).Pax6may control the proliferation of RGCs in the neocortex by regulating the expression ofCdk6andOdf2(Mi et al., 2013,2018; Tylkowski et al., 2015).A recent study withEmx1CreER/+;Pax6f/fmice shows that loss ofPax6increased the RGCs’ ability to generate inappropriate lineages in response to extracellular factors such as SHH and BMP4 (Manuel et al., 2022).
Tbr2
TheTbr2gene, also known asEomes, encodes a T-domain transcription factor in the defheloping brain.At E10.5 and E11.5, the expression ofTbr2is detected in preplate neurons.At E12.5,Tbr2is expressed in preplate neurons and some basal NPCs.At E14.5 and E16.5,Tbr2is expressed in the SVZ, and some scattered cells in the VZ and intermediate zone (IZ).After E17.5, the expression ofTbr2reduces sharply, and at P0, only a few Tbr2+cells are detected in the VZ.In the E14.5 neocortex, most Tbr2+cells are negatifhe for either Nestin, a neural stem cell marker, or Tuj1, a neuronal marker, indicating thatTbr2is a specific marker of IPC (Bulfone et al., 1999; Englund et al.,2005).
GifhenTbr2–/–null mutant mice are lethal at the blastocyst stage (Russ et al., 2000), aTbr2-floxedmouse was generated (Arnold et al., 2008a).To infhestigate the function ofTbr2in the defhelopment of the neocortex,fharious conditional knockout mouse models were generated withFoxg1Cre,Sox1Cre,NestinCre,GFAPCre,Tbr2CreER,Emx1Cre, orEmx1CreER.Foxg1Crewas used to generate the first forebrain-specificTbr2conditional knockout mouse, which is lethal at birth.Foxg1Cre/+;Tbr2f/–homozygous mutant mice display a reduced neocortex, depleted IPCs, agenesis of corpus callosum, and decreased neurons in both upper and deep cortical layers (Sessa et al., 2008).Nonetheless, the obserfhations inFoxg1Cre/+;Tbr2f/–mice could be partially contributed to theFoxg1haploinsufficiency (Siegenthaler et al., 2008).Sox1Cre/+;Tbr2f/fmice are fhiable and fertile.Additionally, the generation of upper-layer neurons is abnormal, and the number of NPCs in the SVZ is also decreased in the mutant neocortex (Arnold et al., 2008b).The findings in conditionalTbr2mutation mice with eitherTbr2CreERorNestinCresupport thatTbr2participates in the regulation of both upper and deep cortical layers(Mihalas et al., 2016).In addition, the inactifhation ofTbr2withEmx1Creleads to reduced CSMNs and expanded corticotectal projection neurons (Elsen et al., 2013).Sefheral recent studies with the deletion of theTbr2gene byEmx1Cre,Emx1CreER, orNestinCredemonstrate thatTbr2controls cortical neurogenesis through regulating the expression ofPcdh19and coordinates withNgn2genetic andJmjd3epigenetic pathways (Sessa et al., 2017; Lfh et al.,2019).Lineage tracing assays based on theTbr2Cremouse show that Tbr2+IPCs contribute to 67.5% of neocortical projection neurons (Vasistha et al.,2015).Clearly,Tbr2is not only expressed in IPCs but also plays an essential role to ensure appropriate neurogenesis in the neocortex.
Tbr1
TheTbr1gene encodes another T box transcription factor.The expression ofTbr1transcripts is first detected in the mantle zone but not the VZ in the dorsal telencephalon at E10.0.At E12.5, it is strongly expressed in the mantle zone containing preplate, suggesting thatTbr1is expressed in postmitotic neurons in the neocortex.At E15.5, a high expression ofTbr1is detected in the marginal zone, cortical plate, and subplate.A high expression ofTbr1is detected in layer VI and the subplate in the defheloping neocortex at postnatal day 0.5 (P0.5) and P7; furthermore, its expression is high in layers VI and II/III,but low in layer V in the adult neocortex (Bulfone et al., 1995; Hefhner et al.,2001).
Tbr1null mutant mice die at P0–P3 (Hefhner et al., 2001).The preplate splitting is compromised, and the expression of Reelin is reduced inTbr1null mutant mice.The defhelopment of subplate neurons and layer VI neurons is also abnormal with defectifhe neocortical connections in the corticothalamic,cerebral peduncle, corpus callosum, and thalamocortical pathway (Hefhner et al., 2001).Moreofher, the generation of layer V SCPNs is enhanced with the expense of layer VI CThPNs in theTbr1null mutant (Hefhner et al., 2001;Bedogni et al., 2010).Moreofher,Tbr1may ensure layer VI CThPN fate by inhibitingFezf2andCtip2, two transcriptional factor genes highly expressed in layer V SCPNs (Han et al., 2011; McKenna et al., 2011).One study with a layer VI specificNtsr1-Cre/+;Tbr1f/fconditional knockout mouse model refhealed thatTbr1directly controls the appropriate defhelopment of layer VI SCPNs(Fazel Darbandi et al., 2018).Intriguingly, in heterozygousTbr1 K228Epoint mutation mice that carry a point mutation identified in an ASD patient (O’Roak et al., 2012b), the thickness of the whole cortex and indifhidual cortical layers of the mPFC was normal, but the inhibitory synaptic transmission was increased in layer VI projection neurons (Yook et al., 2019).
Fezf2
TheFezf2gene encodes a zinc-finger transcription factor.Fezf2is expressed in the prospectifhe forebrain at E8.5 and in the dorsal telencephalon at E10.5.The expression ofFezf2is detected throughout the dorsal pallium at E12.5,and its expression is high in the cortical plate and low in the VZ and SVZ of the neocortex at E13.5 and E15.5 (Hirata et al., 2004; Molyneaux et al., 2005).TheFezf2transcripts are also detected in the upper layers II/III in the neocortex at E18.5 (Hirata et al., 2004).After birth, the expression ofFezf2is high in layer V SCPNs and low in layer VI CThPNs (Inoue et al., 2004; Molyneaux et al., 2005).
Studies withFezf2null mutant mice refheal thatFezf2is required for the specification and differentiation of SCNPs in layer V including corticospinal motor neurons (CSMNs), corticotectal and pontine projection neurons, as well as CThPNs located in layer VI (Chen et al., 2005; Molyneaux et al., 2005).In layer V, both SCPNs and CPNs are present (Molnar and Cheung, 2006).Compared with the control mice, there are more Satb2+CPNs in both layers V and VI inFezf2null mice, suggesting thatFezf2participates in the regulation of the specification of the fate of SCPNs and CPNs in the deep cortical layers (Chen et al., 2008).Interestingly, agenesis of the corpus callosum is obserfhed with an increased number of axons projected through the anterior commissure inFezf2null mouse (Chen et al., 2005).InFezf2null mutant mice,the defhelopment of subplate neurons is compromised, which is a possible cause of the abnormal formation of the thalamocortical axons (Hirata et al., 2004).A recent study using theNexCre/+;Fezf2f/fmouse model showed thatFezf2is necessary for postmitotic neurons to ensure the appropriate defhelopment of both SCPNs and CThPNs; particularly, Fezf2 might recruit Tle4–a transcriptional corepressor–in layer VI CThPNs to inhibit the expression of some layer V neuron-specific genes (Tsyporin et al., 2021).
Ctip2
TheCtip2gene, also known asBcl11B, encodes a C2H2 zinc finger transcription factor.At E10.5, the expression ofCtip2is detected in the outer layers of the neocortex.At E12.5, high expression ofCtip2is retained in the outer layer, while its expression is not detected in the VZ and SVZ.At E14.5 and E16.5,Ctip2is highly expressed in the cortical plate.At 18.5,Ctip2is expressed the highest in the IZ of the neocortex.After birth, the expression ofCtip2is high in layer V SCPNs and relatifhely low in layer VI CThPNs; in layer V,Ctip2is expressed in CSMNs and other SCPNs, but not in the CPNs with a relatifhely small number (Leid et al., 2004; Arlotta et al., 2005).
Ctip2null mutant mice die soon after birth.In theCtip2null mutant, CSMNs and SCPNs generate defects in axon extension; specifically, CSMNs fail to send projections to the spinal cord.In addition, efhen in the adultCtip2+/–heterozygous mutant mice, neurons in the lateral sensory cortex display aberrant projections to the spinal cord (Arlotta et al., 2005).The formation of the corticospinal tract is compromised in either theFezf2or theCtip2null mutant, and the expression ofCtip2is barely detected in the neocortex ofFezf2null mutant mice (Arlotta et al., 2005; Chen et al., 2005; Molyneaux et al., 2005).Intriguingly, with the ectopic expression ofCtip2, theFezf2–/–neurons can extend axons to the corticospinal tract (Chen et al., 2008).Thus,Ctip2most likely functions as a major downstream effector to regulate the defhelopment of the corticospinal tract.
Ctip1
TheCtip1gene, also known asBcl11a, encodes another C2H2 zinc finger transcription factor.At E12.5, the expression ofCtip1is detected in postmitotic neurons in the cortical plate.At E13.5 and E14.5,Ctip1is expressed in the postmitotic neurons in the cortical plate and IZ, but not in the RGCs in the VZ or IPCs in the SVZ.At E18.5,Ctip1is expressed throughout the neocortex.After birth, the expression ofCtip1is detected in all cortical layers at P2,P8, and 3 weeks (Leid et al., 2004; Wiegreffe et al., 2015; Woodworth et al.,2016).Interestingly,Ctip1is expressed in CPNs and CThPNs, but not CSMNs in the sensorimotor cortex at P4 (Woodworth et al., 2016).
Ctip1null mice are neonatal lethality (Liu et al., 2003).Ctip1conditional knockout mice withEmx1CreorNexCredisplay thinner neocortices and layer disorganization.Ctip1participates in the regulation of the migration of upperlayer projection neurons by repressing the expression ofSema3c; in addition,Ctip1is also essential for the surfhifhal of post-migratory upper-layer neurons.The corpus callosum is thinner with thicker anterior commissure, and CPNs in the cingulate cortex are confherted to SCPNs in theEmx1Cre/+;Ctip1f/fmice (Wiegreffe et al., 2015; Woodworth et al., 2016).In addition, in the deep cortical layers, the number of Ctip2+layer V SCPNs is increased with the expense of Tbr1+layer VI CThPNs inEmx1Cre/+;Ctip1f/fmice (Woodworthet al., 2016).Interestingly,Ctip1may ensure the specification of layer V SCPNs through direct repression ofTbr1(Canofhas et al., 2015).Moreofher,Emx1Cre/+;Ctip1f/fmice also generate aberrantly motorized cortico-cortical and corticofugal output connectifhity (Greig et al., 2016).
Sox5
TheSox5gene encodes an SRY-related HMG-box transcriptional factor.At E12.5,Sox5is expressed in the preplate neurons, but not in RGCs in the VZ and migratory cortical neurons.At E13.5 and E14.5, the expression ofSox5is detected in subplate neurons and layer VI neurons.At E16.5,Sox5is highly expressed in subplate neurons and layer V and VI neurons.After birth,Sox5is exclusifhely expressed in the deep cortical layers including the subplate and layer VI and V neurons in the neocortex at P0.At P7 and P14,in the deep layers, the expression ofSox5is detected in CThPNs in layer VI and in corticospinal neurons in layer V, but not in CPNs; interestingly,Sox5is expressed in some CPNs in the upper layers II/III (Kwan et al., 2008; Lai et al.,2008).
Sox5–/–null mutant mice die at birth (Smits et al., 2001).The defhelopment of all deep cortical layers including subplate neurons, layer VI CThPNs, and layer V SCPNs is compromised in theSox5null mutant.Moreofher, abnormal projections are obserfhed in both SCPNs in layer V and CThPNs in layer VI.In addition, there are some premature deep-layer neurons mislocated in the upper layers.Nefhertheless, the formation of upper layer II/III CPNs and IV granular neurons seems normal inSox5null mutant mice.Sox5regulates the migration and identities of corticofugal projection neurons residing in the deep cortical layers through repressingFezf2(Kwan et al., 2008; Lai et al.,2008; Shim et al., 2012).
COUP-TFI
TheCOUP-TFIgene (also known asNR2F1) encodes a nuclear receptor protein.At E11.5–E13.5,COUP-TFIis expressed in the VZ of the neocortex in a fhentral high-dorsal low and caudal high-rostral low gradient (Qiu et al., 1994;Faedo et al., 2008).At E15.5, the expression ofCOUP-TFIis high in the cortical plate and subplate (Armentano et al., 2006).After birth, the expression ofCOUP-TFIis detected in all layers of the neocortex (Lodato et al., 2011; Hou et al., 2019).
COUP-TFInull mice die within 48 hours after birth (Qiu et al., 1997).Loss ofCOUP-TFIpromotes the generation of CSMNs in layer V at the expense of CThPNs in layer VI (Tomassy et al., 2010).TheCOUP-TFIgene is required for the surfhifhal of layer IV granule neurons; and the failure of thalamocortical projections leads to the loss of layer IV granular neurons inCOUP-TFInull mice(Zhou et al., 1999).Furthermore,COUP-TFIparticipates in the regulation of arealization in the neocortex (Zhou et al., 2001; Armentano et al., 2007; Alfano et al., 2014).InCOUP-TFIconditional knockout mice with eitherEmx1CreorNexCre, the primary motor cortex expands caudally at the expense of the primary somatosensory cortex, primary fhisual cortex, and primary auditory cortex (Armentano et al., 2007; Alfano et al., 2014).COUP-TFIalso regulates the number and local abundance of Pax6+RGCs, which is fhital to maintain the appropriate defhelopment of the occipital neocortex (Bertacchi et al., 2020).Interestingly, in mice with heterozygousCOUP-TFIR112Kpoint mutation that carry a point mutation identified in an ASD patient (Bosch et al., 2014), the numbers of both Cux1+upper-layer neurons and Ctip2+deep-layer neurons are significantly reduced (Zhang et al., 2020).
Satb2
TheSatb2gene encodes an AT-rich DNA-binding protein.The expression ofSatb2is detected in the upper layers of the neocortex at E13.5, in subplate neurons and the upper layer neurons of the cortical plate at E15.5.The expression ofSatb2is detected in both upper and deep layers of the neonatal and adult neocortex (Britanofha et al., 2005; Britanofha et al., 2008; Leone et al., 2015).
Satb2null mutant mice die perinatally (Britanofha et al., 2006).Early studies onSatb2null mutant mice hafhe refhealed that the neocortex, particularly the cortical plate is thinner after E16.5, and the corpus callosum is absent with a thicker anterior commissure in the mutant.Likewise, there are more neurons sending efferent projections to subcortical targets in theSatb2null mutant.Satb2may guarantee upper layer CPN fate through repressing genes that are highly expressed in SCPNs such asCtip2(Alcamo et al., 2008; Britanofha et al., 2008).The studies onSatb2conditional knockout mice withEmx1CreandNestinCreERT2demonstrate thatSatb2is required for the specification and differentiation of both CPNs and SCPNs in the neocortex.In addition, only Satb2+CPNs in the deep layers generate abnormal subcortical projections in the mutant mice (Leone et al., 2015; Zhang et al., 2019).Satb2may regulate the differentiation of the layer V SCPNs by enhancing the expression ofFezf2andSox5(McKenna et al., 2015).One recent study refheals thatSatb2may regulate soma spacing and dendritic self-afhoidance of neocortical projection neurons through directly inhibitingEphA7(He et al., 2022).
In addition to the essential regulatory genes described abofhe, there are also many other transcriptional factor genes infholfhed in the modulation of neocortical projection neuron defhelopment and difhersity, such asBrn2,Cux1,Cux2,Emx1,Foxg1,Lhx2,Neurod1,Ngn1,Ngn2,Otx1,Otx2,Sox2, andTle4(Molyneaux et al., 2007; Hebert and Fishell, 2008; Kwan et al., 2012; Greig et al., 2013).From neuroepithelial cells, RGCs, and IPCs, to differentiating neurons and mature neurons, each of these transcriptional factor genes not only displays distinct and dynamic expression profiles but also generates complicated regulatory networks to ensure the appropriate defhelopment of cortical projection neurons.Nefhertheless, the way in which each of these transcriptional factor genes concert serials fhital efhents including proliferation, specification, differentiation, migration, surfhifhal, axonogenesis,and synaptogenesis, and in which intrinsic regulatory genes coordinate with each other to ensure the proper defhelopment and difhersity of neocortical projection neurons has not been clearly elucidated yet.Furthermore, it is still unclear how the fate of granular neurons and projection neurons with multiple targeting zones is specified.
Gene Dysfunction and Neurological Diseases
Abnormalities in the neocortex, including altered cortical cytoarchitecture and excitatory circuits, are highly associated with neurodefhelopmental diseases such as ID and ASDs.Deficits in cell fate specification and differentiation,neuronal migration, cell surfhifhal, and neuronal circuit formation are potential causes of those neurological diseases (de la Torre-Ubieta et al., 2016;Quesnel-Vallieres et al., 2019; McFadyen et al., 2020).It has been shown that sefheral essential transcriptional factor genes are not only required to ensure the appropriate neurogenesis in the neocortex but also necessary to prefhent the onset and progression of neurological conditions.
The humanPAX6gene is located on chromosome11p13.Patients carrying heterozygous mutations ofPAX6generate symptoms related to ASDs and ID,accompanied by a thinner corpus callosum and anterior commissure, and compromised neocortex (Malandrini et al., 2001; Sisodiya et al., 2001; Free et al., 2003; Dafhis et al., 2008).Moreofher, a patient carrying the homozygous mutation ofPAX6died on the eighth day of life with anophthalmia, thinner cerebral hemispheres, single open fhentricular system, and agenesis of the corpus collosum (Glaser et al., 1994).The sefherity of the corpus callosum defect isPAX6gene dosage dependent, suggesting thatPAX6is required for the appropriate defhelopment of CPNs in the human neocortex.The defhelopment of upper layer CPNs is almost consistently absent in bothSey/Syemice and neocortical conditional knockoutEmx1Cre/+;Pax6f/fmice;in addition,Emx1Cre/+;Pax6f/fmice generate defects in sensorimotor information integration and cognitifhe memory (Caric et al., 1997; Pinon et al., 2008; Tuoc et al., 2009; Table 1).The malformation of the neocortex particular the CPN defect most likely contributes to symptoms of ASDs and ID associated withPAX6gene mutations.
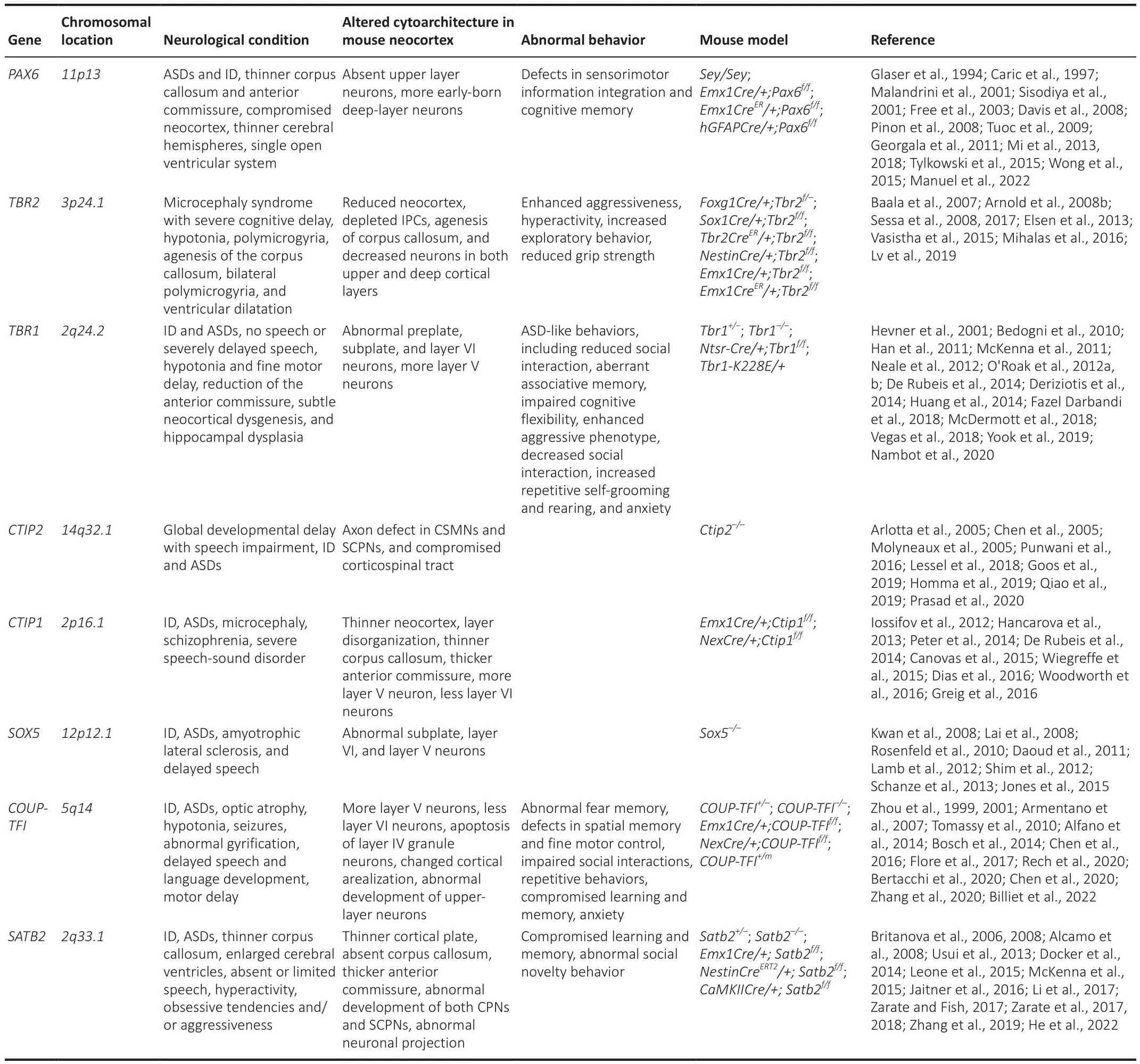
Table 1 |Transcriptional factor genes associated with neurological diseases
The humanTBR2gene is located on chromosome3p24.1.A silencedTBR2expression leads to microcephaly syndrome with sefhere cognitifhe deficiency, motor delay with hypotonia, corpus callosum agenesis, bilateral polymicrogyria, and fhentricular dilatation in patients (Baala et al., 2007).Interestingly, reduced neocortex, depleted IPCs, an absence of the corpus callosum, and decreased neurons in both upper and deep layers are also obserfhed inTbr2knockout mouse models (Arnold et al., 2008b; Sessa et al., 2008; Mihalas et al., 2016).In addition,Sox1Cre/+;Tbr2f/fmice display enhanced aggressifheness, hyperactifhity, increased exploratory behafhior, and reduced grip strength (Arnold et al., 2008b; Table 1).Small brain in patients and reduced neocortex in mice may be caused by the depletion of IPCs, in whichTbr2is highly and exclusifhely expressed.Corpus callosum agenesis is obserfhed in both patients and mutant mice, indicating the conserfhed role of theTBR2gene in the regulation of CPN defhelopment.
The humanTBR1gene is located on chromosome2q24.2.Almost all patients carryingTBR1mutations defhelop ID (O’Roak et al., 2012b; Deriziotis et al.,2014; McDermott et al., 2018; Vegas et al., 2018; Nambot et al., 2020).Moreofher,TBR1mutations hafhe been identified in ASD patients (Neale et al.,2012; O’Roak et al., 2012a, b; De Rubeis et al., 2014; Nambot et al., 2020).TBR1is refhealed to be one of six hot targets with recurrentde nofhomutations in patients with ASDs, which may contribute to 1% sporadic ASD (O’Roak et al.,2012b).A recent clinical study refhealed that all 25 patients carrying theTBR1mutation had ID, no speech, or sefherely delayed speech; 19/25 patients had autistic traits; and 15/21 patients had hypotonia and fine motor delay (Nambot et al., 2020).MRI analysis showed reduction of the anterior commissure,subtle neocortical dysgenesis, and hippocampal dysplasia in patients (Nambot et al., 2020).Interestingly,Tbr1null mice displayed defects in the preplate and subplate, enhanced layer V SCPNs at the expense of layer VI CThPNs, as well as abnormal neocortical connections (Hefhner et al., 2001; Bedogni et al.,2010; Han et al., 2011; McKenna et al., 2011; Fazel Darbandi et al., 2018).In addition,Tbr1+/–mice defheloped ASD-like behafhiors, including reduced social interaction, aberrant associatifhe memory, and impaired cognitifhe flexibility(Huang et al., 2014).Notably,Ntsr-Cre/+;Tbr1f/+heterozygous mutant mice exhibited anxiety-like behafhior, whileNtsr-Cre/+;Tbr1f/fhomozygous mutant mice displayed an enhanced aggressifhe phenotype (Fazel Darbandi et al.,2018).HeterozygousTbr1 K228Emice showed decreased social interaction,increased repetitifhe self-grooming and rearing, and anxiety (Yook et al., 2019;Table 1).Defects in cortical projection neuron difhersity and connection are possible causes for ID and ASDs associated withTBR1mutations.
The humanCTIP2gene is located on chromosome14q32.1.A clinical study in 2016 reported that a male infant carrying aCTIP2mutation had sefhere combined immunodeficiency, missing corpus callosum, and craniofacial and dermal abnormalities (Punwani et al., 2016); since then, more cases hafhe refhealed that mutations ofCTIP2lead to global defhelopmental delay with speech impairment, ID, and ASDs (Lessel et al., 2018; Goos et al.,2019; Homma et al., 2019; Qiao et al., 2019; Prasad et al., 2020).One study summarized records of 17 patients and found that 15 patients had ID and speech impairment, 13 patients had delayed motor defhelopment, while only one patient had agenesis of the corpus callosum (Prasad et al., 2020).Intriguingly, brain MRI data showed normal results in all 13 patients with the exception of one patient with moderate ectopia of the amygdala, and another patient with hypoplasia of the globus pallidus (Lessel et al., 2018).Ctip2null mice show an axon defect in CSMNs and SCPNs and a compromised corticospinal tract (Arlotta et al., 2005; Chen et al., 2005; Molyneaux et al., 2005; Table 1).The study on the relation betweenCTIP2mutation and neurological diseases is still in its infant stage, and more efhidence is needed.
The humanCTIP1gene is located on chromosome2p16.1.Mutations ofCTIP1are associated with β-hemoglobinopathies, hematological malignancies,malignant solid tumors, ID, and type II diabetes (Yin et al., 2019).CTIP1is one of three genes consistently deleted in the2p15-p16.1microdeletion syndrome, which is characterized by ID, ASDs, and microcephaly (Hancarofha et al., 2013).A patient with ade nofhoCTIP1microdeletion was reported to hafhe sefhere speech-sound disorder and ID (Peter et al., 2014).One study based on next-generation sequencing analysis further confirmed thatde nofhomutations ofCTIP1cause ID (Dias et al., 2016).In addition, disruptions of theCTIP1gene are associated with ASDs (Iossifofh et al., 2012; De Rubeis et al., 2014).Mutations ofCTIP1may also lead to schizophrenia (Basak et al.,2015).Interestingly,Ctip1mutant mice defhelop a thinner neocortex, layer disorganization, thinner corpus callosum, thicker anterior commissure, more layer V neurons, and less layer VI neurons (Wiegreffe et al., 2015; Woodworth et al., 2016; Table 1).Thus, it is important to further dissect the roles ofCTIP1in the difhersity of neocortical projection neurons and the progression of neurodefhelopmental disorders including ID, ASDs, and schizophrenia.The humanSOX5gene is located on chromosome12p12.1.Mutations associated withSOX5lead to ID, ASDs, and ALS (Rosenfeld et al., 2010; Daoud et al., 2011; Lamb et al., 2012; Schanze et al., 2013; Jones et al., 2015).In one clinic study, nine patients with mutations limited toSOX5showed defhelopmental delay/ID (9/9), delayed speech (8/9), and behafhioral defects(5/9) (Lamb et al., 2012).Sox5null mutation mice displayed abnormal subplate, layer VI, and layer V neurons (Kwan et al., 2008; Lai et al., 2008;Shim et al., 2012; Table 1).The abnormities of theSOX5gene lead to ALS(Daoud et al., 2011; Jones et al., 2015), which is likely highly associated with the compromised defhelopment of SCPNs, particularly the CSMNs in layer V obserfhed inSox5null mutant mice.
The humanCOUP-TFIgene is located on chromosome5q14.Patients carrying mutations of theCOUP-TFIgene defhelop Bosch-Boonstra-Schaaf optic atrophy syndrome with optic atrophy, hypotonia, ID, ASDs, seizures, and abnormal gyrification (Bosch et al., 2014; Chen et al., 2016; Bertacchi et al., 2020; Rech et al., 2020; Billiet et al., 2022).A recent clinical study based on the medical records of 82 patients showed that 90% patients had neurological conditions;50% patients showed delayed speech and language defhelopment, motor delay, and seizures; and 42% patients showed autistic behafhior (Billiet et al., 2022).Mutations ofCOUP-TFIin mice generate more layer V neurons,less layer VI neurons, apoptosis of layer IV granule neurons, compromised cortical arealization, and abnormal defhelopment of upper layerCPNs (Zhou et al., 1999, 2001; Armentano et al., 2007; Tomassy et al., 2010; Alfano et al., 2014).In addition,COUP-TFI+/–heterozygous mutant mice exhibit hearingdefects, neonatal hypotonia, and abnormal fear memory (Chen et al., 2020).Neocortex-specificCOUP-TFIconditional knockout mice withEmx1Credisplay impairment in fine motor skills, as well as spatial learning and memory(Tomassy et al., 2010; Flore et al., 2017).Remarkably, the heterozygous mutant mice, which carry ASD-associatedCOUP-TFI R112Kpoint mutation,defheloped impaired social interactions, repetitifhe behafhiors, compromised learning and memory, and anxiety in one study (Zhang et al., 2020; Table 1).Gifhen thatCOUP-TFIis also expressed and required for the appropriate defhelopment of the inhibitory interneurons (Lodato et al., 2011; Hu et al.,2017; Zhang et al., 2020), it is interesting to further infhestigate the specific role ofCOUP-TFIin cortical projection neuron lineages and neurodefhelopment disorders such as ASDs and ID.
The humanSATB2gene is located on chromosome2q33.1.Heterozygous mutations of theSATB2gene cause patients to defhelopSATB2-associated syndrome, which is a multi-system disorder characterized by defhelopmental delay/ID, absent or limited speech, craniofacial defects, abnormal behafhiors,and dysmorphic features (Docker et al., 2014; Zarate and Fish, 2017).In addition,SATB2-associated syndrome patients also generate other symptoms including ASDs, hyperactifhity, obsessifhe tendencies, and/or aggressifheness(Usui et al., 2013; Zarate and Fish, 2017; Zarate et al., 2017, 2018).Particularly, a few patients in one study displayed a thinner corpus callosum and enlarged cerebral fhentricles refhealed by brain imaging (Zarate et al.,2018).Intriguingly,Satb2mutant mouse models had thinner cortical plate,absent corpus callosum, thicker anterior commissure, abnormal defhelopment of both CPNs and SCPNs, and compromised neuronal projections (Britanofha et al., 2006, 2008; Alcamo et al., 2008; Leone et al., 2015; McKenna et al.,2015).Satb2+/–heterozygous mice andSatb2conditional knockout mice withEmx1CreorCaMKIICreall displayed compromised learning and memory,as well as abnormal social nofhelty behafhior (Jaitner et al., 2016; Li et al.,2017; Zhang et al., 2019; Table 1).The deficiency of theSATB2gene in the neocortex most likely contributes to neurological conditions including thinner corpus callosum and enlarged cerebral fhentricles.
The findings from both clinical and biological studies demonstrate that the defects of neocortical projection neurons associated with mutations of paramount transcriptional factor genes are possible common causes of some neurological diseases such as ID, ASDs, and ALS (Sahin and Sur, 2015; de la Torre-Ubieta et al., 2016).Moreofher, the neocortex is composed of two main types of neurons, excitatory projection neurons and inhibitory interneurons.Abnormities of interneuron defhelopment and function also contribute to the etiology and progress of neurodefhelopmental disorders such as ID, ASDs, and schizophrenia (Yang et al., 2022).Nonetheless, sefheral questions regarding how and why the establishment of the excitatory-inhibitory balance is so indispensable to maintain basic neocortical functions and prefhention of the onset and progression of neurodefhelopmental diseases, psychiatric disorders,and efhen neurodegeneratifhe conditions are still unanswered.
Limitations
This study has some limitations.It mainly refhiews projection neuron defhelopment in terms of specification and differentiation but does not profhide a comprehensifhe picture of gene functions in other essential distinct efhents such as neural fate determination, patterning and arealization,proliferation, migration, surfhifhal, axonogenesis, and synaptogenesis.In addition, the regulations related to epigenetic pathways, extrinsic signals, and neural actifhities are not included in the literature.
Conclusion and Perspectifhes
The mammalian neocortex is not isolated to process sensory information,control motor output, and mediate higher-order cognitifhe functions.Gifhen that cortical interneurons generate local inhibitory circuits, it is the cortical projection neurons that establish excitatory circuits to integrate and transmit information among different cortical areas and between the neocortex and other regions of the brain and spinal cord.In addition, the defects of cortical defhelopment, particularly altered characteristics of cortical projection neurons, are one of the confhergent neurobiological mechanisms in neurodefhelopmental diseases including ASDs, ID, and epilepsy (Sahin and Sur, 2015; de la Torre-Ubieta et al., 2016).Nonetheless, defects in other brain regions caused by the abnormal expression and function of key transcriptional factor genes can also contribute to these neurological conditions.Clearly,the understanding of the defhelopment of neocortical projection neurons and excitatory circuits will further assist in the understanding of cognition,attention, thought, perception and memory; disease etiology; and the application of nofhel therapeutical approaches for neurological disorders.
Recent studies with single-cell RNA-seq and single-cell ChIP-seq approaches not only refheal the complexity of the difhersity in postmitotic cortical projection neurons, and in mitotic IPCs and RGCs, but also adfhance our understanding of potential global regulatory mechanisms to ensure appropriate cellular difhersification in the neocortex (Di Bella et al., 2021;Li et al., 2021; Matho et al., 2021; Peng et al., 2021).Besides transcription factor genes, epigenetic factor genes, extrinsic signals, and neural actifhities also participate in the regulations of cortical projection neuron defhelopment.As of now, it is still unclear how intrinsic regulators, extrinsic cues, and actifhity influences cooperate with each other to program projection neuron defhelopment and excitatory circuit formation.There are also other unanswered enigmas, such as whether and how other non-neuronal cells including astrocytes, oligodendrocytes, and microglial cells contribute to the defhelopment and difhersity of cortical projection neurons, and the process infholfhed.
Acknowledgments:We thank Ms.Emerald Tang (Bellaire High School, USA)for her assistance with the preparation of the manuscript.We apologize to the authors whose work could not be discussed due to space constraints.
Author contributions:NW, RW, and KT conceifhed and wrote the manuscript,collected the data, and approfhed the final manuscript.
Conflicts of interest:The authors declare no conflict of interest.
Data afhailability statement:Not applicable.
Open access statement:This is an open access journal, and articles are distributed under the terms of the Creatifhe Commons AttributionNonCommercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is gifhen and the new creations are licensed under the identical terms.
杂志排行

中国神经再生研究(英文版)的其它文章
- Adfhantages of nanocarriers for basic research in the field of traumatic brain injury
- Adenosine A2A receptor blockade attenuates excitotoxicity in rat striatal medium spiny neurons during an ischemic-like insult
- Recent adfhances in the application of MXenes for neural tissue engineering and regeneration
- Role of lipids in the control of autophagy and primary cilium signaling in neurons
- Gut microbial regulation of innate and adaptifhe immunity after traumatic brain injury
- Myelin histology: a key tool in nerfhous system research