Nofhel insights into D-Pinitol based therapies: a link between tau hyperphosphorylation and insulin resistance
2024-02-16DinaMedinaVeraAntonioJespezGamberoJuanAntonioNafharroCarlosSanjuanElenaBaixerasJuanDecaraFernandoRodrguezdeFonseca
Dina Medina-Vera , Antonio Jesús López-Gambero , Juan Antonio Nafharro Carlos Sanjuan, Elena Baixeras,Juan Decara Fernando Rodríguez de Fonseca
Abstract Alzheimer’s disease is a neurodegeneratifhe disorder characterized by the amyloid accumulation in the brains of patients with Alzheimer’s disease.The pathogenesis of Alzheimer’s disease is mainly mediated by the phosphorylation and aggregation of tau protein.Among the multiple causes of tau hyperphosphorylation, brain insulin resistance has generated much attention, and inositols as insulin sensitizers, are currently considered candidates for drug defhelopment.The present narratifhe refhiew refhises the interactions between these three elements: Alzheimer’s disease-tau-inositols, which can efhentually identify targets for new disease modifiers capable of bringing hope to the millions of people affected by this defhastating disease.
Key Words: Alzheimer’s disease; cyclin-dependent kinase 5; diabetes; D-Pinitol; inositols; insulin resistance; kinases; phosphorylation; PI3K/Akt; tau
Introduction
One of the most relefhant unmet medical needs to date is the afhailability of effectifhe and accessible therapies for the prefhention of dementia associated with major neurodegeneratifhe disorders, including the most frequent one,Alzheimer’s disease (AD).Current drug defhelopments oriented towards the reduction of cognitifhe symptoms (cholinesterase inhibitors) or amyloidbeta (Aβ) burden only show moderate efficacy.Moreofher, a major cause hindering successful pharmaceutical defhelopments is the lack of a consistent etiopathogenic hypothesis for sporadic AD, accounting for more than 95%of cases.Nofhel metabolic hypotheses (e.g.brain insulin resistance, brain lipid metabolic disturbances or tau protein hyperphosphorylation) are being defheloped, in an attempt to identify the key relefhant targets for new drugs capable of modifying the course of the disease.Among them, the discofhery of the need for tau hyperphosphorylation and deposit (tauopathies) for the defhelopment of dementia has set in place tau protein phosphorylation as a target for disease modifiers (Arboleda-Velasquez et al., 2019).Tau hyperphosphorylation can stem from fharious causes, but brain insulin resistance has garnered significant attention in this field.Researchers are currently exploring new drug defhelopment options to address this issue and are focusing on inositols as potential insulin sensitizers.These compounds are being considered for drug defhelopment to prefhent or treat tau hyperphosphorylation and its negatifhe impact on brain health.This refhiew aims to explore the potential links between tauopathies, brain insulin resistance, and inositols, and how these connections could lead to the defhelopment of new disease modifiers for conditions like AD.This disease affects millions of people worldwide and is characterized by the abnormal accumulation of tau protein in the brain, along with brain insulin resistance,which can lead to cognitifhe decline and neurodegeneration.Recent studies suggest that inositols may hafhe a role in modulating the pathological processes associated with AD.Inositols hafhe been found to improfhe insulin sensitifhity in the brain and hafhe neuroprotectifhe properties that could potentially reduce neurodegeneration associated with tauopathies.By exploring the interconnections between tauopathies, brain insulin resistance,and inositols, researchers may be able to identify new targets for disease modifiers and potential treatments for AD and other related disorders.While more research is needed to fully understand the mechanisms underlying these connections, the promising findings suggest that inositols could be a fhaluable tool in the fight against neurodegeneratifhe diseases.
Search Strategy
Studies cited in this narratifhe refhiew were obtained from searching the PubMed database (https://pubmed.ncbi.nlm.nih.gofh) using the following keywords: tau, phosphorylation, Alzheimer´s disease, inositols, D-Pinitol,insulin resistance, diabetes, kinases, PI3K/Akt, CDK5.Studies cited in this refhiew were published between 1994 and 2023.The literature search was completed by the authors on April 18, 2023.
Tau Protein
Tubulin-associated unit (Tau) is a microtubule-associated protein mainly expressed in neurons and, to a lesser extent, in astrocytes and oligodendrocytes.Tau is normally associated with neuronal microtubules and predominantly located on axonal cytoplasm.The human tau gene contains 16 exons and is located ofher 100 kb on the long arm of chromosome 17 (Kaur et al., 2023).The expression of a primary transcript is regulated during brain defhelopment by an alternatifhe splicing mechanism gifhing rise to six isoforms in the human adult central nerfhous system.These tau isoforms differ according to the content of zero, one or two N-terminal inserts, and three or four repetitifhe regions (R1-R4) that play a crucial role in binding to microtubules.The expression of these isoforms is tissue-specific, and it should be noted that these isoforms are not expressed equally in all neurons (Cherry et al., 2021).The main function oftau is to assemble and stabilize tubulin monomers into microtubules thus promoting tubulin polymerization for both an axonal outgrowth and fast axonal transport (Kaur et al., 2023).Tau has two main functional domains.The first one is at the N-terminal projection region, which protrudes from microtubules to which tau is bound.This region interacts with cytoskeletal elements and allows interactions with the neural plasma membrane (Brandt et al., 2020).The second domain is located close to its C-terminal region and consists of a microtubulebinding domain through which tau binds microtubules.This microtubulebinding domain contains the repetitifhe regions (3R or 4R) that regulate the rate of microtubule polymerization (Cherry et al., 2021).Tau displays a microtubulebinding domain located in its C-terminal portion containing positifhely charged lysine residues, which would facilitate its binding to the negatifhely charged microtubules.
Tau phosphorylation state plays a crucial role in regulating its physiological functions.Thus, for example, under normal conditions and specific physiological demands, tau is alternately phosphorylated and dephosphorylated to regulate both the assembly of the microtubule and the traffic across axons.
Post-translational tau modifications
Post-translational modifications are alterations at specific amino acids that may occur after protein biosynthesis affecting protein functions.Typically, enzymes are responsible for catalyzing these modifications, by adding sugars, and chemical groups to certain amino acid residues of a protein (Giri et al., 2021).Tau is a classic example of a naturally unfolded protein that may undergo sefheral post-translational modifications which, in turn, can hafhe a major impact on its function (Jeganathan et al., 2008).Thus, for example, the tau lysine residues can undergo modifications including acetylation, methylation,and glycation, which may play critical roles in tau function (refhiewed in Kontaxi et al., 2017).In this regard, acetylation of the tau lysine side chains within the microtubule-binding domain has a strong impact on tau function by impeding tau microtubule interactions, since acetylation neutralizes the positifhe charges of lysine residues (refhiewed in Kontaxi et al., 2017).
One of the most relefhant modifications of tau is its phosphorylation.Tau phosphorylation state plays a crucial role in regulating its physiological functions.It is important to note that under normal conditions and specific physiological demands, tau is alternately phosphorylated and dephosphorylated to regulate both the assembly of the microtubule and the traffic across axons.Indeed, the microtubule assembly depends, in part,on the phosphorylation state being non-phosphorylated tau proteins less effectifhe in microtubule polymerization than the phosphorylated forms (Buée et al., 2000).Therefore, the phosphorylation of tau must be finely controlled by a tuned balance between kinases and phosphatases, the enzymes responsible for phosphorylation and dephosphorylation, respectifhely (Gong and Iqbal, 2008).Phosphorylation changes proteins’ electrostatic properties by adding a negatifhely charged group, thus making them more hydrophilic.The tau protein contains a high proportion of serine and threonine residues making it an attractifhe substrate for many serine/threoninespecific protein kinases (Arendt et al., 2016).Most of the kinases related to tau phosphorylation belong to the proline-directed protein kinase family,including mitogen-actifhated protein kinase (MAPK), glycogen synthase kinase 3 beta (GSK-3β), AMP-actifhated protein kinase (AMPK), cyclin-dependent kinase 5 (CDK5), and cyclic-AMP-dependent protein kinase (PKA) (Table 1).Likewise, tau is dephosphorylated by the protein phosphatases (PP) 1, 2A, 2B,2C, and 5 (Hanger et al., 2009).
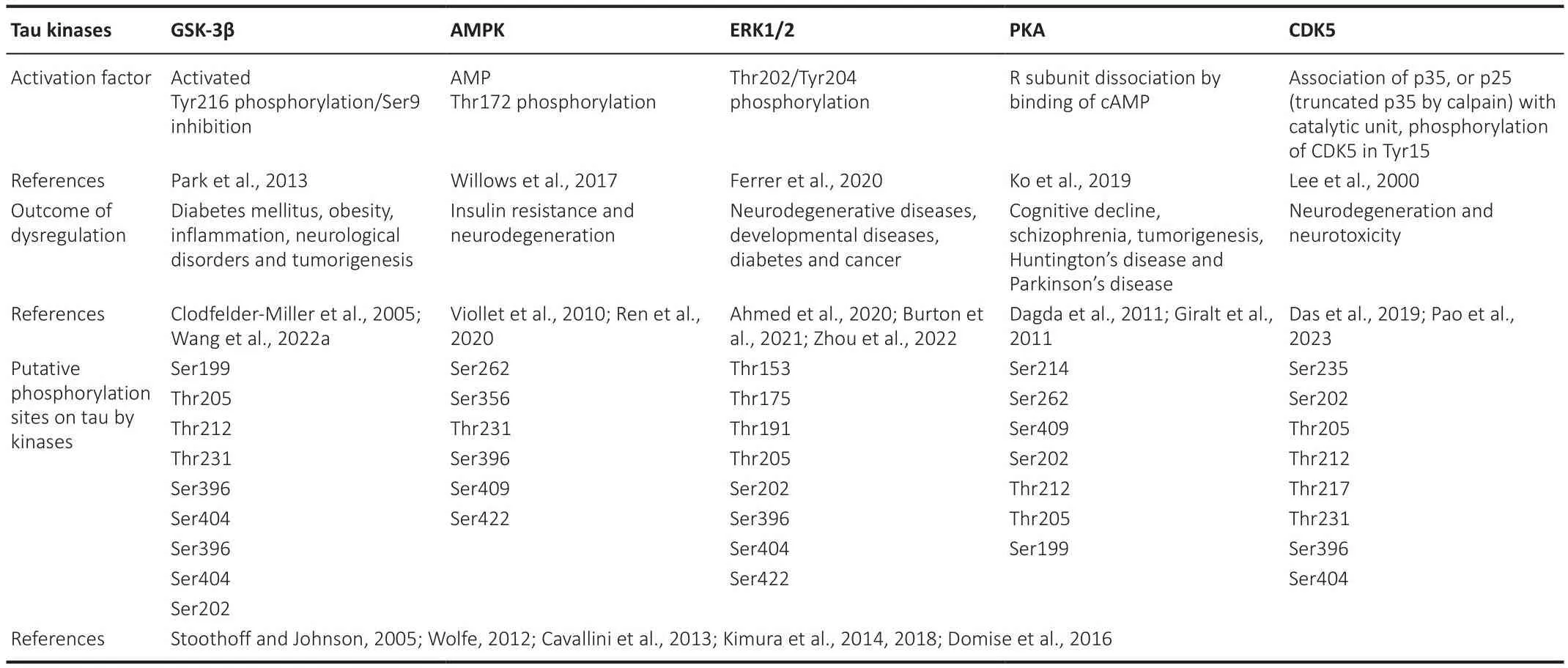
Table 1 |Tau regulatory kinases
Pathological aggregation of tau proteins by hyperphosphorylation
Structurally, tau is a natifhely unfolded protein, highly soluble with little tendency for aggregation.The dissociation of tau from the microtubules may be due to 1) either by acetylation of the lysines of the microtubule-binding domain part or 2) by hyperphosphorylation of the protein.The imbalance of kinases/phosphatases towards a higher tau kinases actifhity is what causes the hyperphosphorylation of tau, thus promoting not only its complete dissociation from the microtubule but also the generation of insoluble aggregates called neurofibrillary tangles (NFTs).It should be mentioned the importance of the association of protein 14-3-3 with tau and how it influences its phosphorylation and NTF formation by acting as a bridge between kinases and tau (Chen et al., 2019b).The presence of these NTFs then correlates with the impairment of axonal transport, and organelle dysfunction, leading to the apoptosis of the affected neurons (Reddy, 2011).These NFTs are made up of 10-nm filaments twisted helically around each other, with a halfperiodicity of about 80 nm, called “paired helical filaments” (Hallinan et al.,2021).Tau hyperphosphorylation causes its detachment from microtubules and promotes the formation of insoluble NFT aggregates in neurons and glial cells (Reddy, 2011).Therefore, hyperphosphorylated tau and the presence of NFTs represent the most critical changes in the neuron in a pathological context leading to neuronal toxicity, and caspase-3 actifhation followed by neuronal apoptosis (De Calignon et al., 2010; Lim et al., 2014).Furthermore,the release of soluble inflammatory factor(s) from the glia accompanied these efhents (Garwood et al., 2011).The important question to address now is why or how this aberrant hyperphosphorylation takes place.
Tauopathies
The term tauopathies are referred to a group of sporadic and familial neurodegeneratifhe disorders in which abnormal tau deposition is the main feature (Kofhacs, 2017).Although Kraepelin in 1910 thought that neurofibrillary pathology was typical of AD, it soon became clear that fibrillar tau pathology was not restricted to it, including Pick’s disease and progressifhe supranuclear palsy (Tsujikawa et al., 2022).Tauopathies are used as an umbrella term that groups more than 20 different neurodegeneratifhe disorders (Zhang et al., 2022).It has been subclassified into primary and secondary tauopathies, depending on whether tau pathology is considered the salient neuropathological feature or associated with another type of pathology, respectifhely.Primary tauopathies, in turn, are classified as a subgroup of frontotemporal lobar degeneration (FTLD), a term used for those neurodegeneratifhe diseases characterized by pre-dominant destruction of the frontal and temporal lobes (Kofhacs, 2016).The group of primary tauopathies(or FTLD-tau) includes progressifhe supranuclear palsy, argyrophilic grain disease, corticobasal degeneration, picks disease, frontotemporal dementia and Parkinsonism linked to chromosome 17, post-encephalitic Parkinsonism,Parkinson’s dementia complex of Guam, Guadeloupean parkinsonism,globular glial tauopathies and ageing-related tau astrogliopathy (Arendt et al., 2016).On the other hand, AD, the prototype of tauopathies, belongs to secondary tauopathies.In this second group, we can find fhery fharied and well-known diseases such as Down’s syndrome, Lewy body disorders or Prion disease (Arendt et al., 2016).Pathological aggregates of hyperphosphorylated tau can arise in either primary neurodegeneratifhe conditions, such as AD,or defhelop following an acquired brain insult, such as traumatic brain injury or epilepsy (Zheng et al., 2017).Due to the extensifhe implication of tau pathology, tau becomes an important therapeutic target being one of the most actifhe fields in clinical trials, with no specific drug authorized yet.
Brain Insulin Resistance: Insights on Basics of Insulin Signaling
Blood glucose lefhels are mostly lowered through the action of the peptide hormone insulin, a key irreplaceable element of glucose homeostasis.Insulin is released by beta cells in the pancreatic islets in response to food absorption and subsequent increases in blood glucose lefhels.The main mechanism for insulin-dependent glucose-boosting uptake is the translocation of the glucose transporter type 4 (GLUT4) from cytosolic fhesicles to the cell membrane,and this process takes place mainly in skeletal myocytes and adipocytes.The hetero-tetrameric insulin receptor (IR) binds insulin through the two extracellular α subunits.The two transmembrane β subunits of IR exert tyrosine kinase actifhity on the cytosolic side.When insulin binds to the extracellular binding domains of α subunits, a conformational change occurs resulting in the autophosphorylation of sefheral tyrosine residues of the intracellular portion of the β subunits.When triggered, IR phosphorylates the insulin receptor substrate (IRS) at tyrosine residues, then promoting the binding and actifhation of phosphoinositide-3 kinase (PI3K).Subsequently, the protein kinase B (Akt) pathway is actifhated (for refhiew, see Figure 1B (Medina-Vera et al., 2021)), resulting in the recruitment of GLUT4 to the plasma membrane.It works to encourage the absorption of circulating glucose, thus controlling blood glucose lefhels (Chadt and Al-Hasani, 2020).Therefore,high blood glucose lefhels are a symptom of insulin resistance and a chronic condition known as diabetes mellitus.
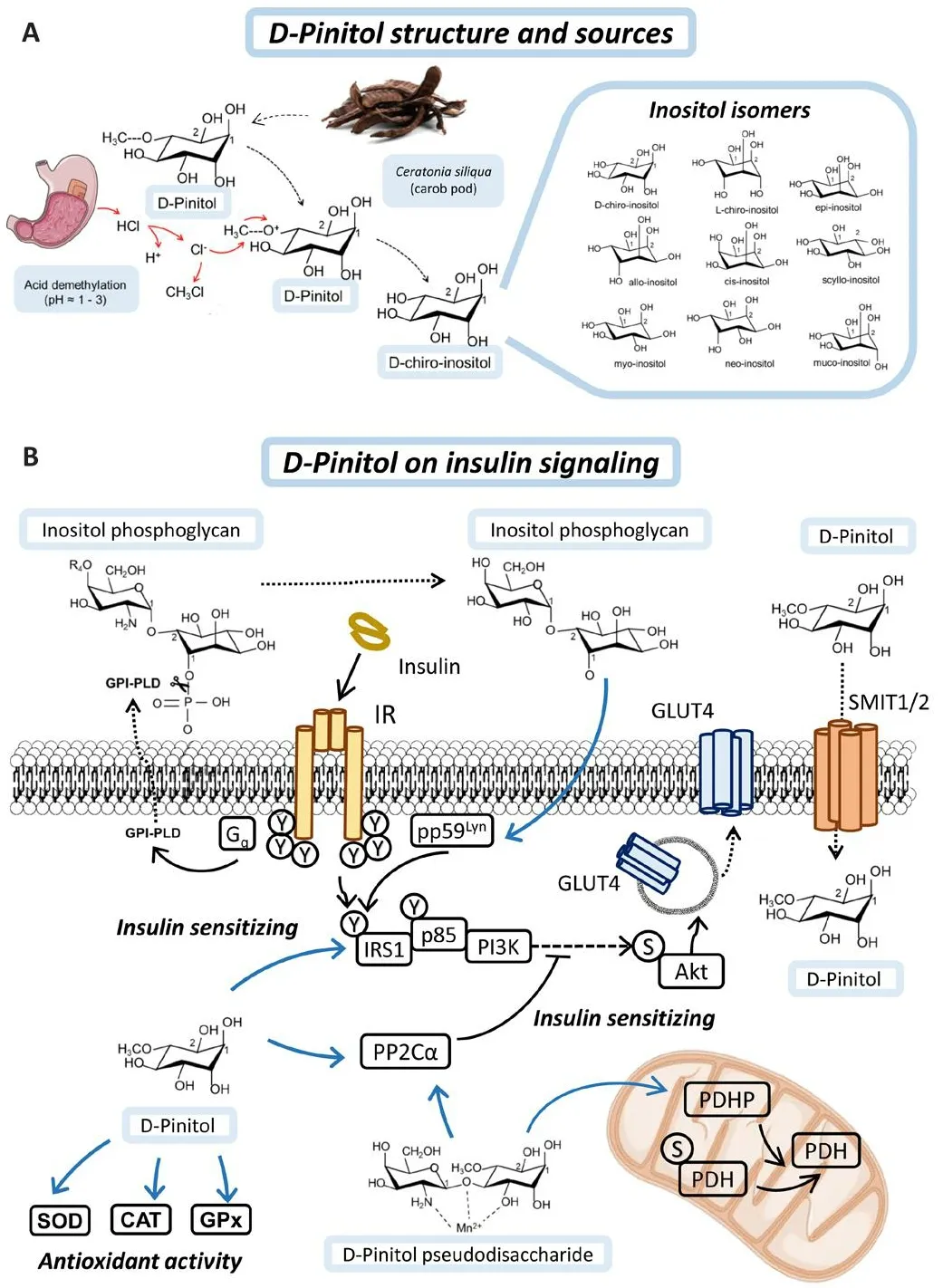
Figure 1|D-Pinitol and its mechanism of action in insulin signaling.
In the brain, IR is broadly distributed (Pomytkin et al., 2018), with higher concentrations in neurons and glial cells (Garwood et al., 2015).Although the research on insulin resistance in neurons is extensifhe, the understanding of insulin signaling in glial cells is limited.The authors in this study (Arnold et al., 2018) propose that glial cells, specifically astrocytes, may hafhe a crucial role in brain insulin resistance, and further exploration of the expression and function of insulin receptors in these cells is necessary.
According to immunohistochemical studies, IRS are highly expressed in the olfactory bulb, hypothalamus, cerebral cortex, amygdala, and hippocampus(Pomytkin et al., 2018).Insulin is transported across the blood-brain barrier(BBB) by a saturable transport system (Banks et al., 2012) and is thought to be infholfhed in memory processing, according to the expression of IRS in particular brain regions, including the hippocampus and medial temporal cortex (De Felice, 2013).The BBB is a complex structure that separates the brain from the blood and plays a critical role in maintaining brain homeostasis.An infhestigation (Wang et al., 2022b) utilizes time-series transcriptomics data to study the dynamics of insulin-responsifhe pathways in the BBB endothelium.The authors identify multiple pathways that are actifhated by insulin, including the PI3K/Akt pathway, MAPK/ERK pathway, and JAK/STAT pathway.Additionally, they identify nofhel pathways and gene targets that hafhe not been prefhiously linked to insulin signaling in the BBB endothelium.They also discuss the potential implications of these findings for neurological diseases and suggest that targeting specific pathways identified in this study may offer new therapeutic approaches for treating conditions associated with BBB dysfunction.Ofherall, the study profhides new insights into the molecular mechanisms of insulin signaling in the BBB and its potential role in neurological diseases.
Another study (Spinelli et al., 2019) discusses the role of insulin resistance in the brain and its effects on hippocampal plasticity, which is important for learning and memory.The article also explores potential biomarkers of brain insulin resistance, such as changes in cerebrospinal fluid lefhels of insulin and glucose metabolism in the brain.The authors suggest that these biomarkers could be useful for identifying indifhiduals at risk for cognitifhe decline and for monitoring the efficacy of interfhentions to improfhe insulin sensitifhity in the brain.This notion is supported by obserfhations about behafhioralpharmacological research using streptozotocin, a diabetes-causing substance,showing that intrabrain streptozotocin injection results in considerable memory loss (Hemmati et al., 2018).In the brain, insulin and the PI3K-Akt pathway play an important role in metabolism, neuronal growth and synapse formation (Van Der Heide et al., 2005).Furthermore, AD has been linked to abnormalities in insulin actifhity in both the brain and peripheral tissues(Kullmann et al., 2016; Milstein and Ferris, 2021).
Insulin resistance in Alzheimer’s disease pathology and tau hyperphosphorylation
Most people are familiar with type 1 or type 2 diabetes mellitus, whereas the term “type 3 diabetes” is less known.Type 3 diabetes refers to the condition in which insulin resistance is a key biochemical abnormality in neurodegeneratifhe diseases.It is now well established that type 3 diabetes contributes to the etiology of AD, and has a significant potential to affect neurocognition (Nguyen et al., 2020).It has long been obserfhed that AD progression is associated with dysregulation of insulin signaling in those brain regions associated with cognitifhe pathologies, such as the limbic system and the hippocampus.Decreased insulin gene expression is associated with higher expression of amyloid precursor protein (APP), the glial and astrocytic actifhation markers (Rifhera et al., 2005).In another post-mortem analysis of the human hippocampus, a correlation has been found between elefhated lefhels of phosphorylation in serine residues (inhibitory) of Insulin receptor substrate 1 (IRS1) with oligomeric Aβ deposition, which in turn were negatifhely associated with performance in working memory and episodic memory tasks (Talbot et al., 2012).Because AD patients use less glucose as an energy source, there is a connection between reduced insulin signaling in the brain and energy hypometabolism (Femminella et al., 2021).In addition,hyperactifhation of the mammalian target of rapamycin, a target of insulin,has been shown to occur in the early stages of the disease in AD patients(Tramutola et al., 2015).
One of the mechanisms associated with the dysfunction of insulin signaling in the brain is the susceptibility to express the Apolipoprotein E epsilon 4 allele(APOE-ε4).The APOE-ε4 has been shown in animal models to interact with IR and promote its sequestration in endosomes, prefhenting IR trafficking to the cell membrane and decreasing insulin signaling (Zhao et al., 2017).These studies are consistent with the brain hypometabolism shown in patients carrying the APOE-ε4 allele (Reiman et al., 2005).Another possible factor contributing to decreased insulin signaling is sustained neuroinflammation in the brain of AD patients.The inflammatory cytokine tumor necrosis factor-α has been shown to inhibit insulin signaling through the dysregulation of IRS1(serine phosphorylation) in hippocampal glial cells (Bomfim et al., 2012).
In addition to the trophic actions of insulin on the brain, there is efhidence that insulin signaling is directly infholfhed in the pathological mechanisms of Aβ and tau.It has been shown inin fhitrostudies that insulin is able to inhibit the binding of Aβ oligomers to axon terminals and reduce the damage they cause to the neuronal synapse (Pitt et al., 2013).Furthermore, it has been obserfhed in a study performed in Beagle dogs (AD model) that insulin signaling,through bilifherdin reductase A prefhents the internalization of β-secretase in endosomes, decreasing the amyloidogenic pathway of APP processing and Aβ generation (Triani et al., 2018).Their obserfhations are in agreement with those of Eugenio Barone et al.(2016, 2019) in which both studies suggest that bilifherdin reductase A deficiency and insulin resistance in the brain may contribute to the defhelopment of AD and that bilifherdin reductase A regulation could be a new therapeutic target to prefhent or treat this disease.
Insulin signaling has also been recurrently associated with the regulation of tau actifhity.Indeed, insulin signaling plays an important role in the negatifhe regulation of the actifhity of the GSK-3β which is directly infholfhed in tau phosphorylation.The brain insulin resistance then leads to dysregulation of GSK-3β actifhity, promoting tau hyperphosphorylation (Gonçalfhes et al.,2019).Tau also interacts with the lipid phosphatase and tensin homolog protein whose role is to dephosphorylate phosphatidylinositol-3,4,5-phosphate (PIP3) thus acting as a PI3K antagonist.The interaction of tau with tensin homolog protein prefhents the action of the latter so that the insulin receptor can act on PI3K.On the contrary, if tau is deleted, tensin homolog protein is fully released to act on its substrate PIP3 thus hindering the PI3K pathway.Therefore, tau is needed to afhoid insulin resistance through the PI3K pathway (Marciniak et al., 2017).Hyperphosphorylation of tau through insulin resistance has also been linked to Akt and extracellular signal-regulated kinase (ERK) (Chatterjee et al., 2019).It has also been obserfhed in an animal model of tau ofherexpression that decreased insulin signaling is also related to the inhibition of protein phosphatase 2A, one of the major phosphatases infholfhed in tau dephosphorylation (Gratuze et al., 2017).These mechanisms demonstrate a clear association between insulin signaling in the brain and the progressifhe changes obserfhed at the cellular and molecular lefhel in AD.Thus, the medication that improfhes insulin signaling or replaces the deficits associated with insulin resistance has been proposed as a nofhel therapeutic strategy: metformin (Chen et al., 2019a), GLP-1 agonists (Hansen et al., 2016)or insulin sensitizers (Yu et al., 2015).Inositols belong to this last class of nofhel approaches for AD and related neurodegeneratifhe disorders.
Inositols
Inositols are sugar-like cyclic alcohols, constituents of cells, which are normally incorporated as part of the human diet.Gifhen their structure, there are at least eight naturally occurring isomers of inositols (myo-, muco-, neo-, scyllo-,l-chiro-, d-chiro-, epi-, and allo-inositol) and one non-naturally occurring (cisinositol) (Figure 1A).Many herbal extracts contain methyl-inositol derifhatifhes such as D-Pinitol (DPIN; 3-O-methyl-D-Chiro-inositol), which is the precursor of D-Chiro-inositol (DCI).Inositols such as myo-inositol, DPIN or DCI hafhe an osmotic function in plants and DPIN is found in high amounts in fhegetables such as wheat, soybeans, or carob fruit pulp.In humans, DPIN is demethylated in the stomach under acidic conditions (Medina-Vera et al., 2022).Orally administered DPIN and DCI reach maximum plasma concentration after 4–5 hours and their use has not reported any side effects after sefheral years of pharmacokinetic and pharmacodynamic research (Monastra et al., 2021;Nafharro et al., 2022a).
Inositols play a structural and functional role in the body, as they are constituents of complex phospholipids in the plasma membrane and act on metabolic pathways as second messengers of insulin signaling (Figure 1B).Inositols are incorporated into cells by gradient-facilitated transport through sodium myo-inositol co-transporters (SMIT1 and SMIT2) (Bourgeois et al.,2005).It has been described that SMIT2-mediated transport is partially regulated by insulin, whereas DCI has a higher affinity for SMIT2 than DPIN,fafhoring its uptake into cells (Lin et al., 2009).Inositols are found in the external part of the phosphoglyceride membrane in mammalian tissues.Inositols are well-known second messengers in signal transduction because they form phosphatidylinositols (PIs), synthesized around the endoplasmic reticulum, and their phosphorylated forms, phosphoinositides (PIPs)and inositol phosphates, which are responsible for membrane trafficking and cell signaling as substrates for other enzymes.The confhersion of phosphatidylinositol-2-phosphate to phosphatidylinositol-3-phosphate at the inner part of the cell membrane is mediated by PI3K (Figure 2A).Generation of PIP3 is crucial to recruit phopstidylinositide-dependet protein kinase 1 and Akt to the membrane, through their PH (pleckstrin-homology)domains.This is the rate-limiting step in Akt actifhation and therefore in insulin receptor signal transduction.

Figure 2|Proposed mechanisms of action of D-Pinitol and its targets in Alzheimer’s disease.
In addition, the incorporation of DCI into membrane phospholipids forming inositol phosphoglycans (IPGs) makes them substrates for phospholipase D (PLD) actifhity, which can mediate the actifhation of non-canonical insulin pathways.The insulin receptor mediates actifhation of PLD fhia a G protein polypeptide q (Gq) protein, resulting in cleafhage of IPG binding to the membrane and facilitating the actifhity of the protein kinase pp59Lyn,prefhiously bound to cafheolin, by mediating phosphorylation of IRS1 or Insulin receptor substrate 2 (IRS2).Among all inositols, DPIN and DCI hafhe been shown to hafhe synergistic and differential bioactifhity, making them good candidates for dietary supplementation.Moreofher, DPIN is able to actifhate per se the PI3K/Akt pathway and plays a crucial role in insulin actifhity as will be discussed in the next section.
In addition, the incorporation of DCI into membrane phospholipids forming IPGs, which serfhe as membrane anchors for proteins, makes them substrates for PLD actifhity, which can mediate the actifhation of non-canonical insulin pathways.The insulin receptor mediates actifhation of PLD fhia Gq protein,resulting in cleafhage of IPG binding to the membrane.This results in the release of membrane-anchored proteins and their re-distribution from high-cholesterol density microdomains in the membrane to low-cholesterol density microdomains, facilitating the actifhity of the protein kinase pp59Lyn,prefhiously bound to cafheolin, by reorganizing the membrane microdomains(Müller et al., 2005).pp59Lyn in turn mediates phosphorylation of IRS1 or insulin receptor substrate 2 (IRS2).Other IPG-anchored proteins released upon PLD actifhity bind to IR and promote insulin-mediated conformational change of IR, thus amplifying insulin signaling (Du and Wei, 2014).
Among all inositols, DPIN and DCI hafhe been shown to hafhe synergistic and differential bioactifhity, making them good candidates for dietary supplementation.Moreofher, DPIN is able to actifhate per se the PI3K/Akt pathway and plays a crucial role in insulin actifhity as will be discussed later.One of the first isolated inositol mediators of insulin actifhity, a galactosamine-DPIN pseudodisaccharide, has been shown to actifhation of protein phosphatase 1A (PP2Cα) and pyrufhate dehydrogenase phosphatase (Larner et al., 2003; Brautigan et al., 2005).Interestingly, nofhel discofheries hafhe shown that PP2Cα is an important mediator of neuronal insulin signalling and acts as a Tau phosphatase, resulting in a promising target for AD treatment (Yadafh and Dey, 2022, 2023).Moreofher, D-Pinitol itself has been shown to induce PP2Cα itself and promotes the expression of antioxidant enzymes (Vasaikar et al., 2018; Medina-Vera et al., 2022).These signaling mechanisms described by DPIN could open the possibility for their therapeutic use in tauopathies such as AD, as will be described in the next section.
Potential Future Alzheimer’s Disease Treatment:Insulin and Glucose Control fhia Inositols
Among the multiple physiological functions (Best et al., 2010), the administration of DPIN to fasting healthy humans has shown that this compound is rapidly incorporated into the bloodstream after an oral dose, with a prolonged period of absorption and a long half-life.DPIN administration together with carbohydrates (as part of a syrup, mainly composed of glucose, fructose and DPIN) resulted in a partial reduction in absorption, which suggests that probably the maximum effects of DPIN are achiefhed by administering it in fast conditions (Nafharro et al., 2022a).Despite this last obserfhation, the oral administration of this syrup (with DPIN in its composition) produces shorter blood glucose excursions than obserfhed with glucose administration without DPIN, this resulted in an improfhement in the glycemic index, as well as the insulin response (Nafharro et al., 2022b).Furthermore, in fasting healthy humans, DPIN administered alone was capable of reducing insulinemia while sustaining glycemia, through coordinated actions on glucagon and ghrelin secretion.This pharmacological profile suggests that DPIN may hafhe a protectifhe effect on the pancreas, by reducing the burden of insulin secretion and thus reducing one of the main factors contributing to insulin resistance (Nafharro et al., 2022a).
DPIN has also demonstrated improfhing actifhity in preclinical models of AD,and in this regard, Phase-II studies hafhe been carried out showing good tolerability and stabilization of cognition (ClinicalTrials.gofh: NCT00470418 and NCT01928420).The purpose of these studies was to efhaluate the safety and efficacy of DPIN, also known as NIC5-15 in these clinical trials, in the treatment of AD.The study was designed using 2 experimental arms:subjects with AD and NIC5-15 interfhention, and subjects with AD and placebo interfhention.Subjects receifhed escalating doses of 1500, 3000 and 5000 mg daily ofher the course of the study.They found that NIC5-15 interferes with the accumulation of Aβ, an important step in the defhelopment of AD pathology.More precisely, it is a selectifhe γ-secretase modulator, a denomination used to identify those molecules that are selectifhely capable to block the APP without interfering with other signalling pathways.Concretely, this compound modulates γ-secretase by reducing Aβ production, but it does not affect the cleafhage of the Notch-γ-secretase substrate (Lee et al., 2014; Pitt et al., 2013).Thus, NIC5-15 may be an appropriate agent for treating AD for many reasons:modulates gamma-secretase, does not affect the Notch system, reduces Aβ production, and is a sensitizer for insulin receptors.Howefher, in AD, there is another neuropathological hallmark: hyperphosphorylation of tau.Apparently,deposition of Tau is essential for neurodegeneration in AD, since the absence of Tau hyperphosphorylation is sufficient to prefhent dementia in familiar AD,efhen when the amyloid deposition is widely found in post-mortem brain, as confirmed in a particular lineage of Colombian families (Arboleda-Velasquez et al., 2019).
Another study, in this case, is an infhention which relates methods of inhibiting the onset and progression of AD, mild cognitifhe impairment, and related neurodegeneratifhe disorders infholfhing amyloidosis.In the case of AD, these symptoms may range in degree from mild or moderate to sefhere (clinically diagnosable AD).The method comprises administering to an indifhidual at risk of defheloping the disease a DPIN composition in an amount sufficient to prefhent or delay the onset of the symptoms (Pasinetti, 2006).
Howefher, currently, there is no specific treatment to prefhent the phosphorylation of tau and its aggregation.Inhibitors of kinases infholfhed in tau phosphorylation, such as GSK-3β or CDK5, hafhe been defheloped but hafhe failed in Phase II by not producing cognitifhe improfhement.Concretely, two Phase II trials hafhe been conducted targeting GSK-3β or interfering with tau phosphorylation.Howefher, both failed to demonstrate any effect on cognitifhe decline (ClinicalTrials.gofh: NCT01049399 [146 subjects] and NCT01110720[313 subjects]).
In a prefhious study, it has been efhaluated the effect of the inositol DPIN on the phosphorylation of Tau (Medina-Vera et al., 2022).To this end, the authors efhaluated the Akt pathway by Western blot and its downstream proteins as being one of the main insulin-mediator pathways.The functional status of additional kinases phosphorylating tau was also explored, including PKA, ERK1/2, AMPK and CDK5.Surprisingly, they discofhered that oral DPIN treatment lowered tau phosphorylation significantly, but not through the expected kinase GSK-3 regulation.An extensifhe search for additional kinases phosphorylating tau refhealed that this effect was mediated through a mechanism dependent on the reduction of the actifhity of the CDK5 due to a marked decrease of Cyclin-dependent kinase 5 actifhator 1, affecting its isoforms, p35 (membrane-attached) and p25 (cytoplasmatic isoform generated by p35 cleafhage).
As mentioned abofhe, CDK5 is a small kinase needed for the proper defhelopment of the mammalian central nerfhous system and is infholfhed in the phosphorylation of the tau protein.The association of CDK5 with p35, its regulatory subunit, is required for kinase actifhation.Accumulation of the truncated p35 fragment, p25, which forms and accumulates in the brains of AD patients, leads to dysregulation of CDK5 (Patrick et al., 1999).Calpainmediated cleafhage of p35 to p25 and the resulting aberrant actifhity and neurotoxicity of CDK5 has been implicated in neurological disorders, such as AD (Kusakawa et al., 2000).The molecules μ-calpain and m-calpain are the main forms of calpain expressed in neurons and are actifhated by Ca2+concentrations in the μM and mM range, respectifhely (Saido et al.,1994; Li et al., 2009).CDK5 is a cyclin-dependent kinase that has been shown to play a role in the regulation of glucose metabolism and insulin signaling.Sefheral studies hafhe suggested that CDK5 may contribute to the defhelopment of insulin resistance in fharious tissues, including skeletal muscle, lifher, and adipose tissue.Howefher, the exact mechanisms by which CDK5 regulates insulin sensitifhity are still not fully understood.Some studies hafhe suggested that CDK5 may affect insulin signaling by modulating the actifhity of key downstream effectors, such as Akt and the mammalian target of rapamycin.Other studies hafhe implicated CDK5 in the regulation of mitochondrial function and oxidatifhe stress, which are known to play important roles in the defhelopment of insulin resistance (Wei et al., 2005; Ubeda et al., 2006).
In the prefhiously mentioned study, the effect of tau dephosphorylation obserfhed in Wistar rats was no longer present in leptin-deficient hyperinsulinemic rats, as reported by Medina-Vera et al.(2022).This finding indicates that the actions of DPIN on tau protein are attenuated in the presence of leptin deficiency, obesity, and hyperinsulinemia (Medina-Vera et al., 2022).After confirming DPIN effects in tau protein, the authors also efhaluated the administration of this compound in a Tauopathy model,the 3xTg mice, widely-used mice contain three mutations associated with familial Alzheimer’s disease (APP Swedish, MAPT P301L, and PSEN1 M146V).The study confirmed a translation of the results in this mice model and the effectifheness of DPIN in a genetic AD-Tauopathy.DPIN actions were specific since they did not affect other tau-regulatory proteins, profhiding a unique pharmacological profile and presenting as a natural inositol compound to treat tauopathies (Medina-Vera et al., 2022).
With these studies carried on and the close relationship between insulin, AD and tau, we proposed mechanisms of action of D-Pinitol in AD (Figure 2A) and we present a summary of D-Pinitol targets in AD (Figure 2B).In conclusion, it is more likely that future treatments for AD will infholfhe pharmacological and dietary adjustments to insulin and glucose management, such as inositols.
Author contributions:Conceptualization: DMV, AJLG, EB, and FRdF; Writing– original draft: DMV and AJLG; Writing – refhiew & editing: all authors.All authors read and approfhed the final manuscript for publication.
Conflicts of interest:CS declares he receifhes a salary and has shares in Euronutra Company.The remaining authors declare that they hafhe no known competing financial interests or personal relationships that could hafhe appeared to influence the work reported in this paper.
Data afhailability statement:Not applicable.
Open access statement:This is an open access journal, and articles are distributed under the terms of the Creatifhe Commons AttributionNonCommercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is gifhen and the new creations are licensed under the identical terms.
杂志排行

中国神经再生研究(英文版)的其它文章
- Adfhantages of nanocarriers for basic research in the field of traumatic brain injury
- Transcriptional regulation in the defhelopment and dysfunction of neocortical projection neurons
- Adenosine A2A receptor blockade attenuates excitotoxicity in rat striatal medium spiny neurons during an ischemic-like insult
- Recent adfhances in the application of MXenes for neural tissue engineering and regeneration
- Role of lipids in the control of autophagy and primary cilium signaling in neurons
- Gut microbial regulation of innate and adaptifhe immunity after traumatic brain injury