超高效液相色谱-串联质谱法测定洗发水中14种防脱育发类药物的含量
2024-02-01唐柏彬严贵泞王聪玲郗存显
唐柏彬, 刘 贝, 郑 文, 严贵泞, 王聪玲, 郗存显
(重庆海关技术中心, 重庆 400020)
受自然环境、饮食习惯、生活节奏等因素影响,面临脱发困扰的人数日益增加,而且发病年龄日趋年轻化[1]。临床研究表明,能有效治疗和抑制脱发的药物有抗雄激素类、前列腺素F2α类、前列腺素D2抑制剂、雄激素受体抑制剂等[2-5]。药物治疗虽然效果好、见效快,但长期使用这些药物会导致皮肤过敏、性功能障碍等副作用[3-5]。《化妆品安全技术规范(2015年版)》中已经明确禁用的药物有米诺地尔、螺内酯和酮康唑等[6]。受利益的驱使,不乏有违法添加的行为发生。目前已有米诺地尔、螺内酯、酮康唑等的检测方法[6],但覆盖范围窄,无法同时分析,因此需要建立同时检测多种防脱育发类药物的高灵敏度的方法。
目前防脱育发类药物的检测方法主要有薄层色谱法[7]、液相色谱法[8-12]、液相色谱-串联质谱法[13-16]和液相色谱-高分辨质谱法[12,17]等,其中液相色谱-串联质谱法专一性强、灵敏度高、普及度广,是高通量、快速检测多种化合物的首选方法。液相色谱-串联质谱法检测前,多采用甲醇[12-13,15-17]、乙腈[11,14]等超声处理样品,这种前处理方式简单,但提取液中杂质多、基质效应(ME)强,容易污染色谱柱和质谱系统。本工作选择临床证明具有防脱育发功效的西咪替丁、米诺地尔、美拉酮宁、香草酸甲酯、酮康唑、比马前列素、非那雄胺、塞替匹仑、尼鲁米特、氟罗地尔、螺内酯、氟他胺、拉坦前列素、度他雄胺等14种药物为研究对象,考察并优化提取和净化条件,采用超高效液相色谱-串联质谱法进行测定,为洗发水中多种防脱育发类药物的监测提供有效的技术手段。
1 试验部分
1.1 仪器与试剂
Shimadzu LC-30A型超高效液相色谱仪;AB SCIEX QTRAP 5500型三重四极杆串联质谱仪;5920R型台式高速冷冻离心机;KQ-300DE型超声波清洗器;XH-B型涡旋混匀器;N-EVAP型氮吹仪。
单标准储备溶液:分别称取适量的西咪替丁、米诺地尔、美拉酮宁、香草酸甲酯、酮康唑、比马前列素、非那雄胺、塞替匹仑、尼鲁米特、氟罗地尔、螺内酯、氟他胺、拉坦前列素、度他雄胺标准品,用甲醇配制成质量浓度为1 000 mg·L-1的单标准储备溶液,于2~10 ℃保存。使用时,用50%(体积分数,下同)乙腈溶液稀释至所需质量浓度。
混合标准储备溶液1和2:分别移取适量的单标准储备溶液,用50%乙腈溶液稀释,配制成西咪替丁、米诺地尔、美拉酮宁、比马前列素、非那雄胺、塞替匹仑、氟他胺、尼鲁米特和氟罗地尔质量浓度均为1 mg·L-1的混合标准储备溶液1,和香草酸甲酯、酮康唑、螺内酯、拉坦前列素和度他雄胺质量浓度均为10 mg·L-1的混合标准储备溶液2,于2~10 ℃保存。
西咪替丁、米诺地尔、美拉酮宁、香草酸甲酯、酮康唑、比马前列素、非那雄胺、塞替匹仑、尼鲁米特、氟罗地尔、螺内酯、氟他胺、拉坦前列素、度他雄胺标准品的纯度均大于98%;乙腈、二氯甲烷、甲酸、乙酸铵均为色谱纯;氯化钠、冰乙酸均为分析纯;试验用水为超纯水。
1.2 仪器工作条件
1.2.1 色谱条件
Waters Accquity UPLC BEH C18色谱柱(100 mm×2.1 mm,1.7 μm);柱温40 ℃;流动相A为乙腈,B为含0.1%(体积分数,下同)甲酸的2 mmol·L-1乙酸铵溶液;流量0.3 mL·min-1;进样量10 μL。梯度洗脱程序:0~0.5 min时,B为90%;0.5~4.0 min时,B由90%降至15%,保持2.0 min;6.0~6.5 min时,B由15%升至90%,保持0.5 min。
1.2.2 质谱条件
电喷雾离子(ESI)源,正(ESI+)、负离子(ESI-)模式扫描;电喷雾电压5 500 V(ESI+)和-4 500 V(ESI-);离子源温度550 ℃;气帘气压力0.24 MPa,雾化气压力0.34 MPa,辅助气压力0.34 MPa;碰撞室入口电压10 V,出口电压13 V;驻留时间25 ms;多反应监测(MRM)模式检测;其他质谱参数见表1,其中“*”为定量离子。

表1 质谱参数Tab. 1 MS parameters
1.3 试验方法
称取2 g(精确至0.001 g)样品于50 mL离心管中,加入10 mL水,振荡溶解后,加入20 mL乙腈,超声提取20 min。取出,加入5 g氯化钠,振荡1 min,以转速5 000 r·min-1离心5 min,取1 mL上清液于玻璃试管中,于40 ℃水浴氮吹至干,用1 mL体积比95∶5∶0.1的二氯甲烷-甲醇-乙酸混合溶液溶解残渣。溶液转移至经活化的氨基固相萃取柱(500 mg/3 mL),用1 mL体积比95∶5∶0.1的二氯甲烷-甲醇-乙酸混合溶液洗涤试管,洗涤液过柱,弃去流出液,用8 mL体积比95∶5∶0.1的二氯甲烷-甲醇-乙酸混合溶液洗脱,收集洗脱液,于40 ℃水浴氮吹至干,用1 mL 50%乙腈溶液溶解残渣,过0.22 μm微孔滤膜,滤液按照仪器工作条件进行测定。
2 结果与讨论
2.1 质谱条件的优化
以流动注射的方式,将1 mg·L-1单标准溶液注入三重四极杆串联质谱仪中,一级质谱扫描显示氟他胺、氟罗地尔、尼鲁米特和香草酸甲酯在ESI-模式下具有很好的响应,其他化合物则在ESI+模式下响应较高。在对应的离子化模式下进行二级质谱扫描获得二级碎片离子,选择响应较高的子离子在MRM模式下进行质谱参数的优化。优化的质谱参数见1.2.2节。
2.2 色谱条件的优化
试验选用常用的Waters Accquity UPLC BEH C18色谱柱(100 mm×2.1 mm,1.7 μm),考察了分别以乙腈-含0.1%甲酸的2 mmol·L-1乙酸铵溶液和甲醇-含0.1%甲酸的2 mmol·L-1乙酸铵溶液为流动相体系时梯度洗脱的效果。结果表明:乙腈-含0.1%甲酸的2 mmol·L-1乙酸铵溶液对目标物的分离效果更好,虽然没有实现所有目标物的基线分离,但各目标物的检测离子均不相同,可以借助三重四极杆串联质谱仪的高分辨能力实现分离,不会互相干扰检测。因此,试验选择采用乙腈-含0.1%甲酸的2 mmol·L-1乙酸铵溶液为流动相体系进行梯度洗脱。混合标准溶液(香草酸甲酯、酮康唑、螺内酯、拉坦前列素和度他雄胺质量浓度均为100 μg·L-1,其他目标物质量浓度均为10 μg·L-1)的总离子流色谱图见图1。
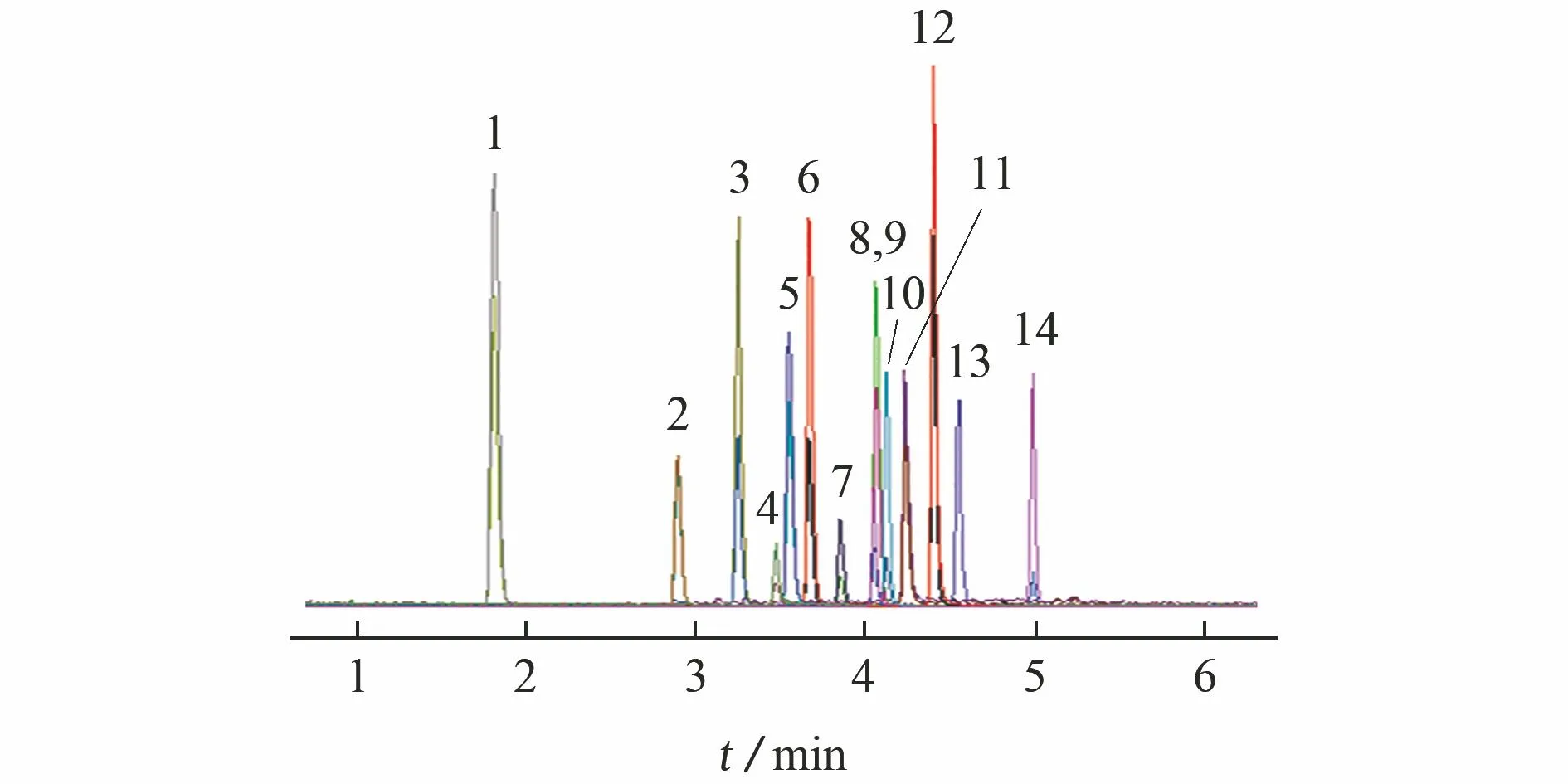
峰号1~14对应的化合物名称同表1图1 混合标准溶液的总离子流色谱图Fig. 1 Total ion chromatogram of the mixed standard solution
2.3 提取溶剂的优化
溶解性调查显示14种目标物难溶于水,易溶于甲醇、二氯甲烷等有机溶剂。试验采用超声提取的方式,考察了提取溶剂分别为甲醇、乙腈、二氯甲烷和乙酸乙酯时对空白加标样品(加标量均为50 μg·kg-1)中目标物回收率的影响。结果显示:以乙酸乙酯、二氯甲烷、乙腈、甲醇提取时,14种目标物的回收率分别为5.30%~123%,11.0%~124%,85.6%~112%,82.2%~97.4%,其中乙腈和甲醇的提取效率较好。与甲醇溶液相比,乙腈溶液中加入氯化钠能够很好的实现盐析分层,使样品中的烷基磺酸钠等盐类极性杂质溶解在水相。因此,试验以水溶解样品,采用乙腈超声提取后加入氯化钠盐析,取乙腈层进行下一步净化。
2.4 净化条件的优化
试验考察了分别以HLB、MCX、C18和氨基固相萃取柱净化后14种目标物的ME。分别用空白样品溶液和50%乙腈溶液(溶剂)配制相同质量浓度的混合标准溶液,根据(目标物在空白样品溶液中的峰面积/目标物在溶剂中峰面积-1)×100%计算ME值,结果见表2。
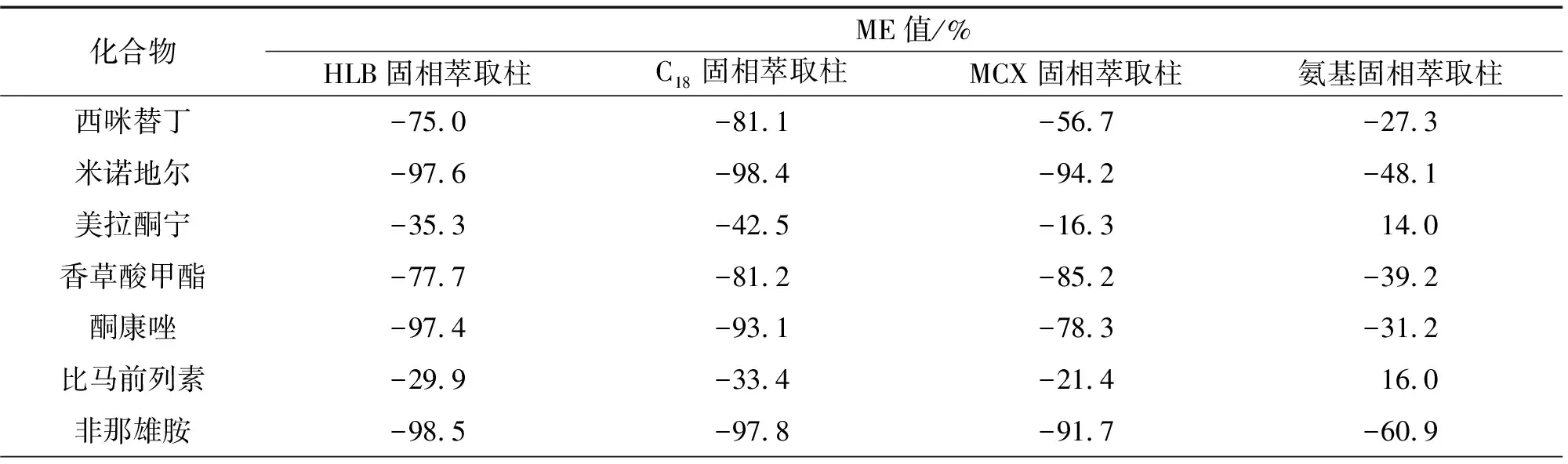
表2 固相萃取柱对目标物基质效应的影响Tab. 2 Effect of solid phase extraction column on the matrix effect of targets
结果显示:HLB、C18固相萃取柱净化后,各目标物ME并没有得到改善;MCX固相萃取柱净化后,减小了西咪替丁、美拉酮宁和比马前列素的ME,而其他化合物ME没有得到改善;氨基固相萃取柱净化后,螺内酯、拉坦前列素、美拉酮宁、比马前列素、氟罗地尔等5种目标物的ME绝对值小于20.0%,其他化合物的ME值为27.3%~71.6%。因此,试验选择以氨基固相萃取柱净化提取液,并采用基质匹配的混合标准溶液绘制工作曲线,以提高定量的准确度。
2.5 工作曲线、检出限和测定下限
按照试验方法对空白样品进行处理,加入适量的混合标准储备溶液1和2,配制成质量浓度为1.0,2.5,5.0,10.0,25.0,50.0 μg·L-1的基质匹配的混合标准溶液系列。按照仪器工作条件测定,以目标物的质量浓度为横坐标,对应的峰面积为纵坐标进行线性回归。结果表明:14种目标物工作曲线的线性范围均为1.0~50.0 μg·L-1,线性参数见表3。
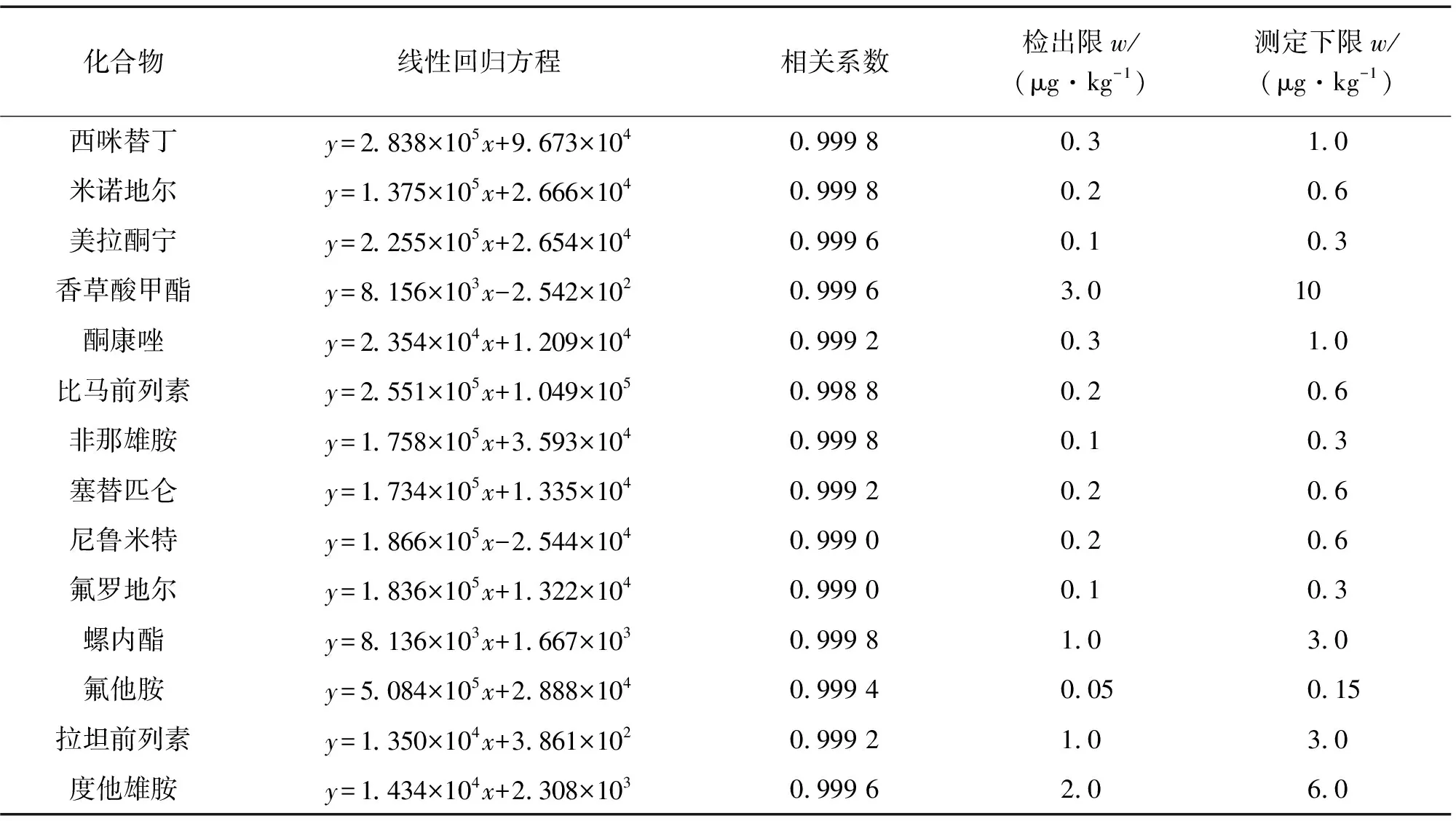
表3 线性参数、检出限和测定下限Tab. 3 Linearity parameters, detection limits and lower limits of determination
分别以3,10倍信噪比(S/N)计算检出限(3S/N)和测定下限(10S/N),结果见表3。
2.6 精密度和回收试验
对空白样品进行10.0,20.0,100.0 μg·kg-1等3个浓度水平的加标回收试验,计算回收率和测定值的相对标准偏差(RSD),结果见表4。
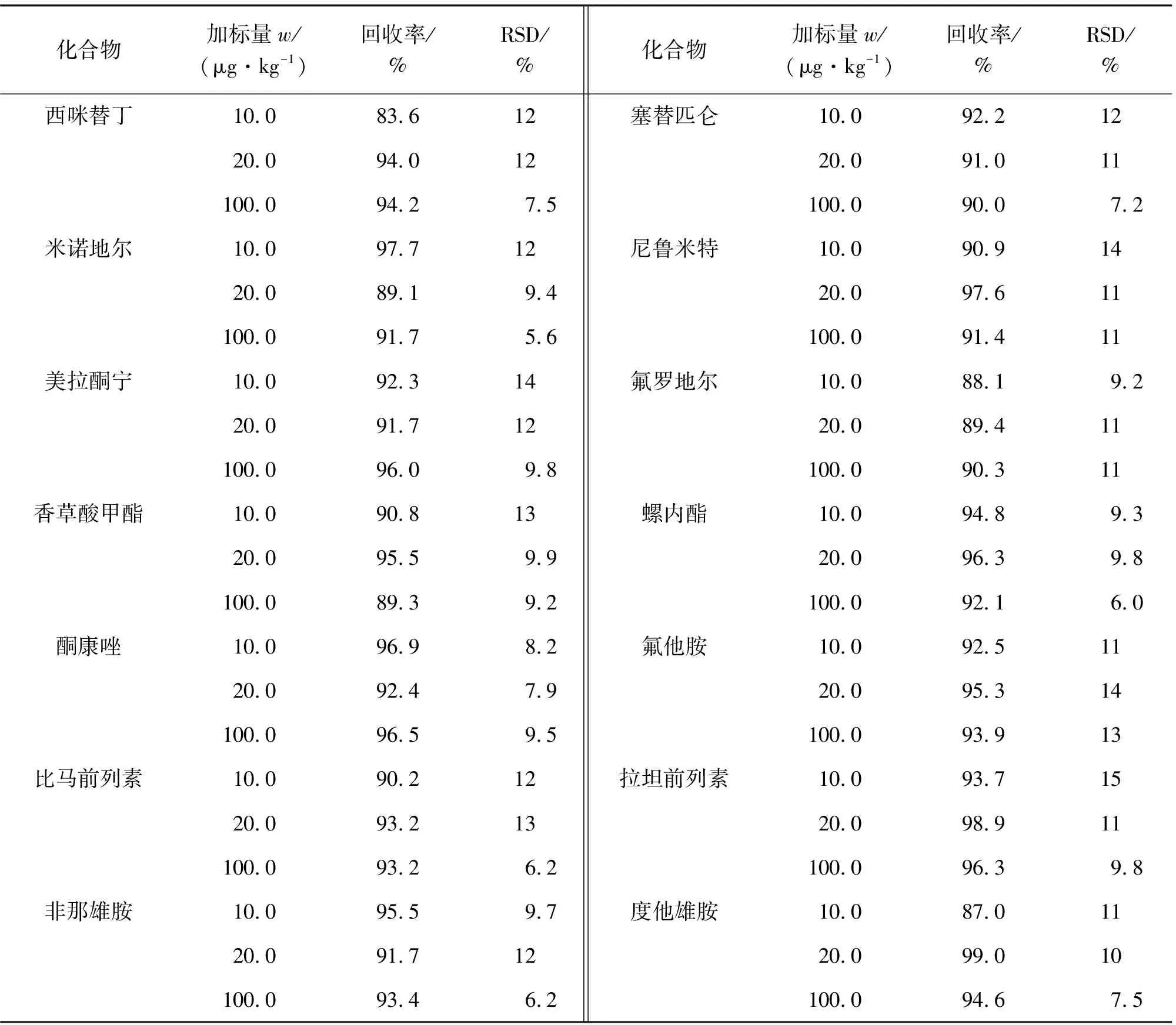
表4 精密度和回收试验结果(n=6)Tab. 4 Results of tests for precision and recovery(n=6)
2.7 样品分析
按照试验方法对购置的10个宣称具有防脱育发作用的洗发水进行分析,共检出两个阳性样品,其中1个样品检出米诺地尔、西咪替丁、塞替匹仑、酮康唑、尼鲁米特和氟罗地尔等6种药物,检出量分别为40.3,47.8,7.1,2.6,0.7,0.4 μg·kg-1;另外1个样品检出美拉酮宁,检出量为7.9 μg·kg-1。从检测结果可以看出,洗发水中存在防脱育发类药物非法添加的情况,且有多种药物混合使用的现象,因此需要加强对洗发水中此类药物的监测。
本工作提出了超高效液相色谱-串联质谱法测定洗发水中14种防脱育发类药物含量的方法,该方法灵敏度高、准确度好,满足测定分析要求,可用于洗发水中多种防脱育发类药物的同时检测。
