Ge/Sn 合金化对CsPbBr3 钙钛矿热载流子弛豫影响的非绝热分子动力学研究
2024-02-01王斐杨振清夏雨虹刘畅林春丹
王斐 杨振清 夏雨虹 刘畅 林春丹
(中国石油大学(北京)理学院,能源交叉学科基础研究中心,油气光学检测技术北京市重点实验室,北京 102249)
1 引言
钙钛矿太阳能电池(PSCs)由于其性能优异、易于合成、成本低等特点,近十几年来一直是光伏领域的研究热点[1–8].钙钛矿的结构通式为ABX3(A为阳离子,B为金属离子,X为卤素基团),自2009 年首次将其用作太阳能电池材料以来,单结钙钛矿太阳能电池效率已从3.8%提升至26.1%,双结全钙钛矿叠层电池效率更是高达28%[9],已然是硅太阳能电池的有力竞争者.其中,全无机铯铅卤化物钙钛矿CsPbX3具有高荧光量子产率[10]和广泛可调的光谱[11],并表现出较高的光稳定性以及优异的热稳定性[12],在众多钙钛矿材料中脱颖而出[13].而在各种CsPbX3钙钛矿中,CsPbBr3具有最大的热稳定性,且基于CsPbBr3的器件在应用中表现出优越的发光特性[14],是光电领域的明星材料.然而,虽然铅基PSCs 效率很高,但含铅导致的毒性以及电池器件长期稳定性较差是其大规模商业化的瓶颈.同时,根据Shockley-Queisser理论,卤化铅钙钛矿的带隙通常不在单结太阳能电池的最佳范围内(1.3—1.4 eV).为了进一步缩小带隙并减少对环境的危害,研究者们主张在钙钛矿B位进行同价替换(如Ca,Bi,Sr,Sn,Ge)来改进PSCs[15,16].其中Sn 和Ge 作为Pb 的同主族元素,对环境友好,且三者的混合钙钛矿均具有直接带隙,受到广泛关注[17,18].目前,在开发这类材料方面已有一些成果,如Liu 等[19]验证了Ge 原子可以有效地保护内部Sn 原子,使其不容易被氧化;Li等[20]对MASn0.5Ge0.5I3进行了电子结构计算以及分子动力学模拟,并与MASnI3比较,表明混合阳离子策略可以同时提高钙钛矿的稳定性和载流子寿命.然而,无铅混合Sn-Ge 钙钛矿虽然能构建高效稳定的钙钛矿太阳能电池,但它们的各方面性能与含铅的同类材料相比仍然缺乏竞争力.在钙钛矿中,激发态载流子动力学过程对其性能的研究至关重要,利用第一性原理对体系基态物理性质进行计算的方法目前已经较为成熟,然而对于激发态性质的描述,尤其是激发态载流子动力学,仍然存在一定的困难.众所周知,非辐射电子-空穴复合缩短了载流子寿命,是激发态电荷能量损失的主要途径[21,22],这直接影响着钙钛矿材料的光电转换效率和电荷传输性能.因此,最大化降低钙钛矿太阳能电池的非辐射复合能量损失是提升电池器件效率的研究重点,对于优化太阳能电池和光电材料具有重要意义.
合金化可以通过多种机制影响载流子的寿命,如带隙调节、杂质能级、晶体结构改变和电子-声子相互作用的调整.这些机制可以单独或联合作用,使合金材料具有更好的光电性能和载流子传输性能.本工作受前人实验及计算工作的启发,利用非绝热分子动力学(NAMD)结合含时密度泛函理论(TDDFT),研究了CsPbBr3以及其合金化结构CsPb0.75Ge0.25Br3和CsPb0.5Ge0.25Sn0.25Br3的激发态载流子动力学过程,最终得到非辐射电子-空穴复合情况和其中减少电荷能量损失的机制,本研究有助于对影响铯基卤化铅钙钛矿性质的关键因素的认识,以寻找性能更优异的钙钛矿材料.
2 计算方法
本文采用含时密度泛函理论结合非绝热分子动力学模拟的方法展开研究.用量子力学描述运动快、质量轻的电子,用经典力学的方法处理运动慢、质量重的原子核.计算过程中,使用VASP[23]软件包进行几何优化、电子结构、绝热分子动力学和非绝热分子动力学计算,用投影缀加波(PAW)方法[24]描述电子-离子相互作用,用广义梯度近似下的Perdew-Burke-Ernzerhof (PBE)泛函处理电子交换相关能[25].其中,结构优化和电子结构计算采用4×4×4 的k点网格,由于结构在Γ 点处有直接带隙,因此仅计算Γ 点处的非绝热耦合.几何优化中原子位置弛豫到所有原子受到的力均低于1×10–5eV/Å,平面波截断能为500 eV.使用Nose-Hoover恒温器将系统温度调节到300 K.随后,在微正则系综中获得以1 fs 为时间步长的3 ps 绝热分子动力学(MD)轨迹.对于载流子的含时演化,利用含时Kohn-Sham 理论中的退相干诱导的表面跳跃(DISH)方法[26],在DISH 算法中加入退相干以反映核轨迹分支并获取正确的量子跃迁时间尺度,退相干时间远短于电子-空穴复合时间,因此在计算中考虑退相干效应,该方法已被广泛用于研究体系中的激发态动力学.整个NAMD 模拟过程借助Zheng 等[27]在2019 年发布的激发态动力学第一性原理计算软件Hefei-NAMD,选择3 ps 绝热MD轨迹的所有几何形状作为电子-空穴复合的NAMD模拟的初始条件,计算电子和空穴在纳秒尺度中的布居演化.
3 计算结果
3.1 主要结果
电子-振动相互作用产生非绝热(NA)电子-声子耦合并诱导量子相干性损失,对应于非弹性和弹性电子-声子散射,它们都影响电子-空穴复合.研究结果表明,CsPbBr3体系退相干时间为8.97 fs,相较于该体系,CsPb0.75Ge0.25Br3体系的退相干时间缩短至6.96 fs,但非绝热耦合增加,它们对电子-空穴复合有相反的作用;而CsPb0.5Ge0.25Sn0.25Br3体系较单掺杂Ge 时进一步减少了电子和空穴波函数的重叠并加快了原子波动,NA 耦合更小且退相干时间缩短至6.20 fs;此外,合金化使得带隙缩小.较小的量子退相干与较小的带隙和较大的NA耦合相竞争,最终,电子空穴复合时间分别延长至1.6 倍以及4.2 倍,遵循CsPb0.5Ge0.25Sn0.25Br3>CsPb0.75Ge0.25Br3> CsPbBr3的顺序,具体数据见表1.模拟结果说明了由于合金化引起的快速量子退相干有效抑制了电子-空穴复合,为高性能钙钛矿材料和器件的设计提供了有意义的见解.

表1 CsPbBr3, CsPb0.75Ge0.25Br3, CsPb0.5Ge0.25 Sn0.25Br3 体系的带隙、非绝热耦合平均绝对值(NAC)、退相干时间(Tpd)和非辐射电荷复合时间(Trec)Table 1.Bandgap,averaged absolute value of NA coupling (NAC),pure-dephasing time (Tpd),and nonradiative charge recombination time (Trec) of CsPbBr3, CsPb0.75Ge0.25Br3, CsPb0.5Ge0.25Sn0.25Br3 systems.
3.2 晶体结构
非绝热耦合(NAC)与模拟体系中的原子数紧密相关,为了排除尺寸效应对光诱导动力学的影响,基于弛豫的CsPbBr3构建了包含40 个原子的2×2×2 的CsPbBr3钙钛矿超晶胞(图1(a)),其立方相包含8 个Cs 原子,8 个Pb 原子以及24 个Br原子,用一个Ge 或Sn 取代一个Pb 对应12.5%的替换浓度.在原始结构基础上,进行合金化,构建了相同尺寸的CsPb0.75Ge0.25Br3以及CsPb0.5Ge0.25Sn0.25Br3体系.在计算前,对所有体系均进行了结构优化达到最稳定状态.CsPbBr3的优化晶格常数为5.900 Å,与实验值5.870 Å一致[28],合金化体系中随着加入原子比例的增大优化晶格常数大小递减.通过300 K 下30 ps 的从头算分子动力学(AIMD)模拟,研究了系统的热稳定性.整个模拟过程中所有体系总能量只有很小的波动,此外,没有观察到结构重建或键断裂发生,表明体系合金化前后在300 K 下结构稳定.

图1 (a) CsPbBr3,(b) CsPb0.75Ge0.25Br3,(c) CsPb0.5Ge0.25Sn0.25Br3 三种钙钛矿体系在0 K(上)和300 K(下)的晶体结构Fig.1.Crystal structure diagrams of three perovskite systems at 0 K (top) and 300 K (bottom) of (a) CsPbBr3,(b) CsPb0.75 Ge0.25Br3,(c) CsPb0.5Ge0.25Sn0.25Br3.
图1 显示了CsPbBr3以及Ge/Sn 合金化后的CsPb0.75Ge0.25Br3和CsPb0.5Ge0.25Sn0.25Br3体系的几何结构,包括0 K 时优化后的结构以及300 K时分子动力学轨迹中选取的有代表性的结构.CsPbBr3的平均Pb—Br 键长为2.984 Å,与实验值2.960 Å[29]一致.温度升高到300 K,CsPbBr3的几何结构只发生了轻微的变形,而合金化体系则出现了明显的畸变,这是因为原始晶胞具有稳定的无机骨架,而合金化体系中引入的Ge 离子和Sn离子比Pb 离子半径小,使得其中的[BBr6]4–(B=Pb/Sn/Ge)八面体的键角变化大于原始体系,以提供更多空间来适应键长变化.我们观察到热波动将3 个体系中的平均Pb—Br 键长延长,延长率分别为0.71%,1.18%及2.93%.相应地,在两个合金化体系中,Ge—Br 平均键长延长率分别为1.76%和3.26%,Sn—Br 平均键长增长率为1.03%.可见,随着Ge/Sn的逐步加入,键长变化逐渐增大,原子位移逐渐增大,退相干速度逐渐加快.Ge,Sn 合金化引起了显著的局部几何畸变,并影响电子振动相互作用,进一步影响电子空穴复合.
3.3 电子性质
图2 为采用优化的几何结构计算的三个体系的投影态密度(PDOS).图2(a)显示,在CsPbBr3中,价带顶(VBM)主要由Br 原子贡献,Pb 原子次之,而导带底(CBM)主要来自Pb 原子的贡献.图2(b),(c)显示,合金化之后,Ge 和Sn 的作用主要体现在CBM 上,对VBM 几乎没有影响,导带底相较原胞显著降低.A位Cs 原子对VBM 和CBM均没有直接贡献,因此,它对NA 电子-声子耦合几乎没有影响,但会通过诱导Pb-Br 八面体的倾斜间接影响电子-空穴复合.且态密度(DOS)在VBM附近相对于CBM 显示出更尖锐和更窄的峰,预测空穴的非辐射寿命比电子短.

图2 (a) CsPbBr3,(b) CsPb0.75Ge0.25Br3,(c) CsPb0.5Ge0.25Sn0.25Br3 体系的DOS 和PDOSFig.2.Projected density of states (PDOS) of (a) CsPbBr3,(b) CsPb0.75Ge0.25Br3,(c) CsPb0.5Ge0.25Sn0.25Br3 systems.
虽然通过自旋轨道耦合校正的HSE06 泛函方法或GW 方法计算的带隙已经与实验取得了良好的一致,但它需要极高的计算成本,因此,选用PBE泛函,计算结果显示,3 个体系均为直接带隙,CsPbBr3的带隙为1.73 eV,与之前第一性原理计算结果一致,但小于实验值2.36 eV,这是由于半局部PBE 泛函高估了电子离域效应,导致计算结果数值偏小.而CsPb0.75Ge0.25Br3及CsPb0.5Ge0.25Sn0.25Br3体系的带隙分别为1.44 eV 和1.05 eV,3 个体系的带隙呈现递减趋势,同上文态密度所述一致.一般来说,带隙越小,越有利于光吸收,可见Ge 和Sn 的加入能够有效减小带隙,增强光吸收能力.另外,带隙的减小会使电子和声子量子更接近共振,从而增强NA 耦合.
3.4 电荷密度
大多数半导体内部的电荷弛豫均发生在(亚)皮秒时间尺度上,因此,可假设电子和空穴在复合之前已经弛豫到带边缘态(LUMO 和HOMO).NA 电子-声子耦合的强度取决于HOMO (最高占据分子轨道)和LUMO (最低未占分子轨道)之间的重叠,它们之间的矩阵表示为〈φj|∇R|φk〉,核速度为 dR/dt[30],通常,电荷密度重叠越大则NA 耦合越强.图3 显示了HOMO 和LUMO 的电荷密度.从图3(a)可以看出,原始体系下,HOMO 处电荷分布在Br 和Pb 原子上,LUMO 处电荷均匀地分布在Pb 原子上,形成自由电荷载流子系统,并促进波函数混合以及NA 耦合的实现,分析结果与PDOS 结果一致.由图3(b),(c)可见,在晶格中引入Ge 和Sn 后,HOMO 和LUMO 上电子和空穴重叠增加,电子和空穴波函数之间的混合程度增加.
3.5 逆参与比
关键电子态的电荷分布有助于深入认识电子-空穴相互作用的强度.为了给电荷密度分析的论点提供更可靠的证据,我们计算了300 K 下3 ps MD 轨迹上VBM 和CBM 的逆参与比(IPR)[31].IPR=N×,0<IPR≤1.其中N为Kohn-Sham 方程中电子态i所占权重,Ki代表电子态i所贡献的电荷密度.通过计算IPR 可以得到电荷在某个状态下波函数的局域性评估,IPR 值越大,电荷分布越局部化,IPR=1 表示完全局域化状态.图4(a),(b)分别为原始体系和两个合金化体系在价带和导带的IPR 序列对比,灰色为原始体系,淡紫色为掺杂体系,深紫色为两者重叠的部分.如图4 所示,与CsPbBr3相比,合金化结构中VBM的IPR 值变化不大,CBM 的IPR 值显著提高.CBM主要由Pb,Ge 以及Sn 原子支持,它们的混合产生了额外的无序.而VBM 主要由Br 原子支持,所受Pb-Ge-Sn 混合的影响不大.该变化可以证明我们的合金化策略在常温下对新的电子状态贡献较大.
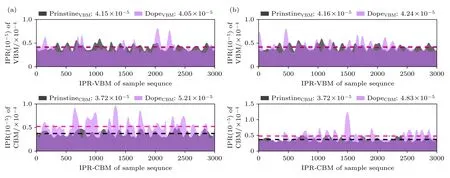
图4 (a) CsPb0.75Ge0.25Br3 与CsPbBr3 以及(b) CsPb0.5Ge0.25Sn0.25Br3 与CsPbBr3 体系的VBM 和CBM 的IPR 对比(虚线为平均值)Fig.4.Comparison of IPRs between VBM and CBM of the doped system and the original system (dashed lines represent the average).
3.6 非辐射弛豫
电子-振动相互作用产生非弹性和弹性电子-声子散射.这两个因素都会影响非辐射电子-空穴复合.非弹性电子-声子散射产生NA 耦合,它通过调节量子跃迁期间声子损失的电子能量,使CBM 上的电子和VBM 上的空穴复合.弹性电子-声子散射会破坏通过NA 耦合在CBM 和VBM 之间形成的叠加,从而导致量子退相干.一般来说,较短的退相干时间会延迟量子动力学.如果退相干时间很小,量子跃迁就会停止,即量子芝诺效应[32].
退相干方法可以观测微观系统的量子态演化,提供了粒子在势能面间跳跃的物理证明,α 态对应的退相干时间为τα:
其中ci是绝热表象下的布居数,rαi是α 态在一次测量坍缩过程中的失相率,在绝热动力学采样当中,核轨迹及各个绝热态的退相过程可以通过态叠加干涉项描述,
其中含时项Jij为两态系统的波包重叠,环境诱导的退相干进程可以被估计,Zheng 等[27]将上式的正则平均近似展开为二阶积累项,
当核轨迹按如上式累积项获得响应函数,拟合得到一元高斯函数的特征时间可记为两态系统的退相时间,
同时核势能在准连续的含时演化中作为系统浴,两态产生的耦合矢量是含时演化哈密顿量的一部分[33].
采用DISH 方法模拟了载流子在价带和导带的捕获和复合过程,得到了整个激发态动力学过程.为了确定电子-空穴复合的时间尺度τ,根据短时近似P(t)=exp(−t/τ)≈1−t/τ,将NAMD 的200 ps数据拟合到指数衰减中,弛豫过程在无限远处达到平衡,将98%的布居平衡视为弛豫结束.电子与空穴的寿命相差1 个数量级,当两者在电池材料两极积累净电荷,形成饱和的漂移电场后,富余的电子会在互相排斥的非弹性散射中湮灭,所以在本研究构造的样本中,首要考虑空穴的布居数演变.图5为3 个体系的空穴在第一激发态居群的演化曲线,电子由导带向价带跃迁,空穴由价带向导带跃迁.表1 给出了3 个体系中电子和空穴在导带和价带间演化的非绝热耦合系数、退相干时间以及电子空穴复合时间等相关数据.可见,与原始体系相比,由于退相干时间缩短,CsPb0.75Ge0.25Br3体系电子-空穴复合时间延长到176 ps,而CsPb0.5Ge0.25Sn0.25Br3体系退相干时间最短,复合时间延长至462 ps.相对于CsPbBr3,合金化体系中退相干时间的缩短是导致电子空穴复合时间延长的关键因素.更小的带隙和更大的NA 耦合与退相干时间变化的影响相反,然而,与退相干时间差异的20%相比,带隙和非绝热耦合的差异较小.

图5 CsPbBr3、CsPb0.75Ge0.25Br3 和CsPb0.5Ge0.25Sn0.25Br3的HOMO 布居数演化Fig.5.Population evolution of HUMO of the pristine CsPb-Br3,CsPb0.75Ge0.25Br3,CsPb0.5Ge0.25Sn0.25Br3.
4 结论
利用非绝热分子动力学结合含时密度泛函理论研究了全无机卤化铅钙钛矿CsPbBr3以及在其B位进行Ge/Sn 合金化后的CsPb0.75Ge0.25Br3和CsPb0.5Ge0.25Sn0.25Br3体系的非辐射电子-空穴复合,并将合金化结构与原始结构进行了比较.计算表明,在CsPbBr3中进行Ge,Sn 合金化会导致明显的几何畸变,从而对电子结构产生显著的影响.另外,带隙缩小了0.29 eV 以及0.68 eV.其次,当较大较重的Pb 被较小较轻的Ge/Sn 取代时,NA耦合增加,但退相干时间显著缩短,总体来说,非辐射复合时间分别延长了1.6 倍以及4.2 倍.本工作的合金化策略对改善钙钛矿太阳能电池材料性能具有一定的促进作用,可以认为B位双原子合金化是相干性调节的有效手段,通过加速退相干,延缓了非辐射电子-空穴复合,从而延长了载流子寿命.这有利于降低太阳能电池中的电荷和能量损失,提高光电转换效率.可见金属阳离子对钙钛矿太阳能电池的性能起着重要作用,为控制和抑制电荷复合、提高光伏效率提供了有效手段.
