角质形成细胞与2型炎症互作在特应性皮炎发病机制中的作用
2024-02-01张睿文黄小宝王芳
张睿文, 黄小宝, 王芳
中山大学附属第一医院,广东 广州 510080
特应性皮炎(atopic dermatitis,AD)是以2型免疫反应为主导的慢性炎症性皮肤病,以皮肤干燥、反复发作的湿疹样皮疹和剧烈瘙痒为基本特征[1]。近年来,AD患病率在全球范围内持续增长,影响15%~20%的儿童[2]及3%~10%的成人[3-4]。可见,AD已成为全球性健康问题,不仅给患者及其家属造成巨大的疾病负担,也给人类健康管理带来沉重的经济与社会负担。
虽然AD的病理生理学机制复杂,涉及多个因素[1],但皮肤屏障功能障碍和2型炎症反应起着主导作用。角质形成细胞(keratinocyte,KC)是构成皮肤屏障的主要成分,屏障破坏使得皮肤对病原微生物及过敏原的通透性增加,在这些物质刺激下,KC通过释放细胞因子和趋化因子,作用于皮肤常驻免疫细胞以及趋化浸润的2型炎症细胞,引起或加剧皮肤2型炎症反应。可见,受损的皮肤屏障是引起皮肤免疫失衡的机制之一。同时,2型炎症细胞释放的2型炎症因子将反作用于KC上相应的受体,促使其结构和/或成分发生进一步改变,加剧皮肤屏障功能的破坏。由此可见,KC与2型炎症之间存在相互关系,可能对AD症状的加重具有重要作用,而打破这二者之间的恶性循环可能是控制AD病情的重要切入点。
瘙痒作为AD的主要症状,常以恶性“瘙痒-搔抓循环”为特征。最新研究表明,2型炎症因子通过激活感觉神经元上的受体介导瘙痒[5],而KC释放的表皮因子除具有激活2型炎症的作用之外,也被证实具有直接致痒效应[6-8]。因此,瘙痒症状可能也是KC与2型炎症相互作用的体现之一。
本文通过阐述皮肤屏障生理学、KC在AD炎症与瘙痒中的作用、2型炎症对KC的影响,以及AD创新药物对皮肤屏障功能的修复作用等,解析KC与皮肤2型炎症的互作关系在AD发病机制中的重要性,以期为AD的研究及治疗提供新思路。
1 皮肤具有屏障作用与感觉功能
皮肤是保护人体免受外界有害成分入侵的第一道屏障,其屏障功能主要由表皮实现。表皮细胞中KC占80%以上,其经历增殖、分化和角化的过程形成结构化表皮。细胞间脂质在KC分化过程中形成,主要包括胆固醇、游离脂肪酸和神经酰胺(ceramide, CER)。其中,CER是维持皮肤屏障功能必不可少的脂类,在减少经表皮水分丢失(transepidermal water loss, TEWL)和保持皮肤水分含量中起重要作用。
不同分化阶段的KC表达不同的角蛋白(keratin, K)。K1和K10是已知正常表皮中KC终末分化的早期标志物,而K6、K16和K17在过度增殖或再生的表皮中表达[9]。在晚期分化过程中,KC可合成丝聚蛋白(filaggrin, FLG)、兜甲蛋白(loricrin, LOR)和内披蛋白(involucrin, IVL)等屏障相关蛋白。FLG是KC的关键结构蛋白,与LOR、IVL共同形成角化包膜,后者是维持角质层细胞形态与功能的重要结构。
除了作为物理屏障与免疫屏障,皮肤也是感觉器官,受周围感觉神经高度支配。感觉神经元在表皮形成表皮内游离神经末梢(intraepidermal free nerve endings, IEFNs)。IEFNs在KC之间传递温度、疼痛和瘙痒等皮肤感觉,但最近的研究表明,KC也可作为机械或化学等刺激的感觉传感器,并直接与IEFNs交流[10-11]。
2 KC在AD皮肤屏障破坏中的作用
临床上用于评价皮肤屏障功能的客观指标主要是皮肤生理指标,如TEWL和水分含量。AD患者表现为皮肤TEWL增加与水分减少,该现象与KC中脂质和屏障蛋白的含量改变相关[12]。AD患者皮肤中脂质的改变主要是神经酰胺的数量和组成变化,研究发现无论是AD患者的皮损还是非皮损部位,神经酰胺谱的组成与健康对照组相比都有显著性差异,且神经酰胺亚类的比值水平与TEWL、表皮水分含量相关,这证明表皮分化改变可引起AD皮肤屏障功能失调[13]。Sho等[14]也发现AD中神经酰胺谱紊乱的现象,并且表明AD缓解期的角质层神经酰胺谱可能是预测后续病情恶化的生物标志物。
较多研究均发现AD患者皮肤中FLG、LOR及IVL等屏障蛋白的表达下调[15-16],这将直接导致皮肤屏障的破坏,成为AD发病机制的关键环节之一。研究证实了FLG突变与婴儿期湿疹和AD有关[17]。KC结构和/或成分的改变最终可导致AD患者皮肤屏障功能缺陷。
3 KC异常加重AD炎症
KC对AD炎症的加重作用主要表现在通过释放表皮因子、炎症因子等直接作用于皮肤免疫细胞,包括2型炎症细胞,如肥大细胞、嗜碱性粒细胞、2型固有淋巴样细胞(type 2 innate lymphoid cell, ILC2)和Th2细胞[18-19],从而启动、活化、加剧皮肤免疫向2型免疫偏移。除此之外,KC释放的表皮因子、酶类等也被发现能够直接激活感觉神经,引起瘙痒。同时,激活的感觉神经通过释放神经肽、神经递质反作用于KC和皮肤免疫细胞,加重免疫反应。因此,KC、2型炎症细胞、感觉神经三者彼此联系,形成AD的神经免疫机制(图1)。
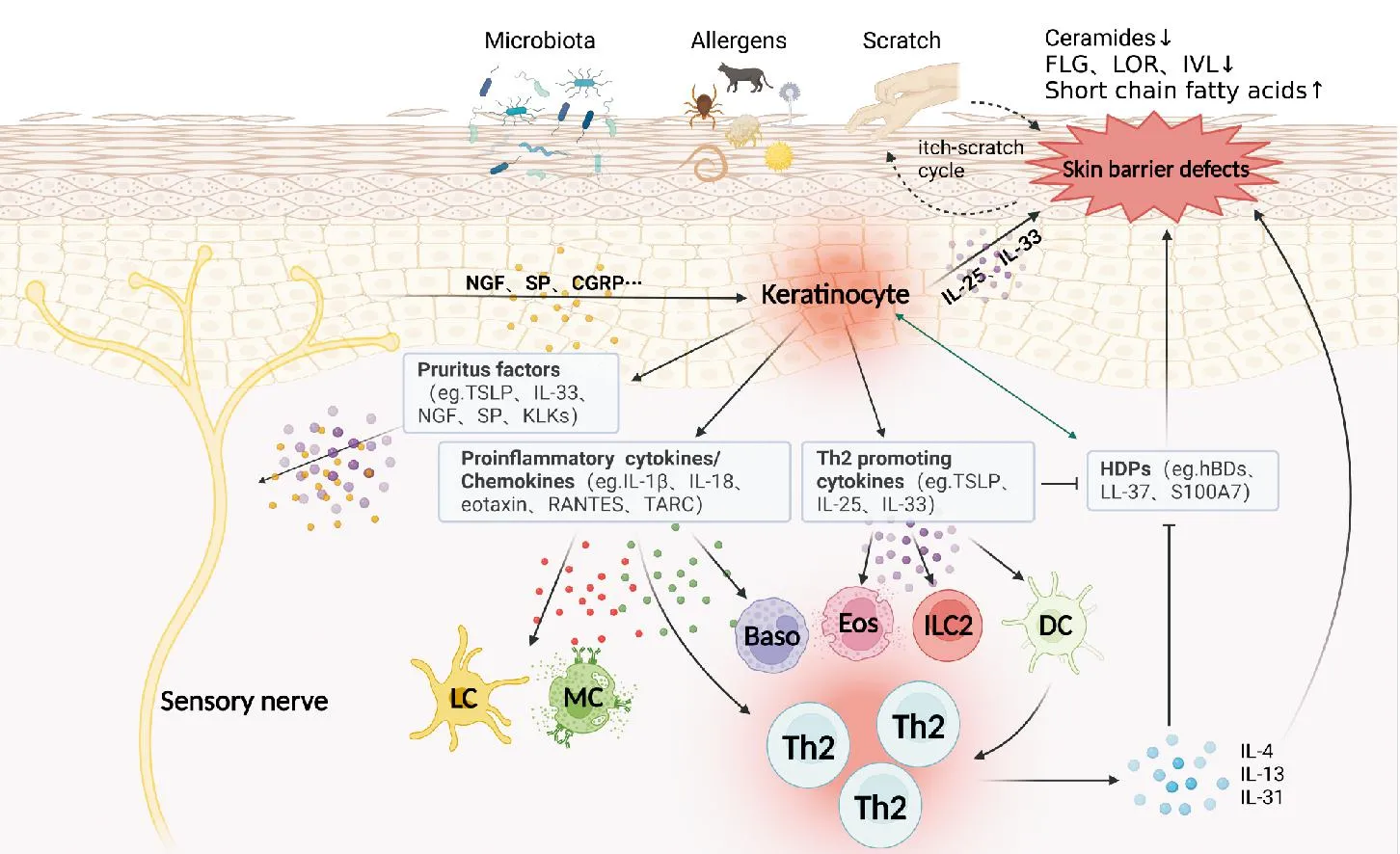
FLG:丝聚蛋白;LOR:兜甲蛋白;IVL:内披蛋白;TSLP:胸腺基质淋巴细胞生成素;RANTES:受激活调节正常T细胞表达和分泌因子;TARC:胸腺和活化调节趋化因子;NGF:神经生长因子;SP:P物质;KLK:激肽释放酶;CGRP:降钙素基因相关肽;HDP:宿主防御肽;hBD:人β-防御素;LC:朗格汉斯细胞;MC:肥大细胞;Baso:嗜碱性粒细胞;Eos:嗜酸性粒细胞;ILC2:2型固有淋巴样细胞;DC:树突状细胞;Th:辅助性T细胞
3.1 表皮因子
在AD患者的皮损中,KC产生的表皮因子(或警报素)主要有胸腺基质淋巴细胞生成素(thymic stromal lymphopoietin, TSLP)、白介素(interleukin, IL)-25和IL-33。TSLP已被证实为变应性炎症的“主开关”[20],其通过树突状细胞诱导原始T细胞向Th2细胞分化,对2型免疫偏倚的诱导起着关键作用。TSLP在AD患者皮肤中的表达与疾病的严重程度、表皮屏障破坏的程度有关[21]。而IL-25通过直接激活ILC2促进IL-13的产生,引起AD病变皮肤中Th2细胞的募集,最终导致2型炎症的发生发展[22]。在皮肤特异性过表达IL-33的转基因小鼠中,可观察到ILC2、肥大细胞和嗜酸性粒细胞浸润的AD样表型[23]。IL-33还可促进KC自身和Th2细胞分别释放TSLP和IL-31,从而放大Th2反应[19]。IL-25和IL-33不仅可以直接促进2型炎症的发生,而且还通过下调FLG的表达加剧皮肤屏障受损,从而在炎症反应和皮肤屏障功能破坏之间建立联系[24-25]。另一方面,KC释放的TSLP可直接作用于感觉神经元诱发强烈的瘙痒[6]。一项小鼠模型研究表明,IL-33通过受体ST2直接激活感觉神经元引发瘙痒,并且可能使神经敏化,以促进慢性瘙痒[7]。
3.2 表皮因子外的炎症因子
携带FLG突变的AD患者中IL-1β和IL-18的水平在角质层中升高,并且与TEWL呈正相关[26]。研究表明IL-1β可能是通过诱导TSLP和减少皮肤屏障蛋白而获得AD表型的早期关键介质[27]。AD病变部位显示多种趋化因子的增加,主要来源于KC,包括趋化因子CC配体2(chemokine C-C ligand, CCL2)/单核细胞趋化蛋白(monocyte chemoattractant protein, MCP)-1、CCL5/受激活调节正常T细胞表达和分泌因子(regulated upon activation normal T-cell expressed and secreted, RANTES)、CCL11/eotaxin、CCL17/胸腺和活化调节趋化因子(thymus and activation-regulated chemokine, TARC)以及CCL26/eotaxin-3等,这些趋化因子可招募和活化固有免疫细胞和T细胞[18],促使其产生细胞因子诱导Th2细胞极化,进一步扩大2型炎症效应。
3.3 其他酶类与肽类物质
激肽释放酶(kallikreins, KLK)是新发现的致痒细胞因子,皮肤中高表达的KLKs主要由KC分泌。Chavarria-Smith等[28]发现AD患者表皮中KLK5和KLK7的表达上调。先前的研究表明,KLK5通过激活蛋白酶激活受体2(protease-activated receptor 2, PAR2)导致瘙痒[29]。最新研究证实KLK7在小鼠AD样皮肤和人类AD皮肤中过表达,且通过非炎症机制促进AD相关的瘙痒[30]。
除了神经系统本身以外,KC也是神经生长因子(nerve growth factor, NGF)的重要来源。一项免疫组化研究观察到,在AD患者KC中有更高水平的NGF,且表皮神经纤维密度较高[31]。Roggenkamp等[32]证实了NGF是KC介导的神经突生长增加的关键参与者。NGF可增加神经纤维释放P物质(substance P, SP)和降钙素基因相关肽(calcitonin gene-related peptide, CGRP)等,从而导致神经源性炎症,如SP能够直接结合肥大细胞上的MRGPRX2受体,引起肥大细胞脱颗粒;SP还可作用于KC,增加IL-1β、IL-6和NGF的产生[33]。CGRP则通过增加IL-4、CCL17和CCL22的产生并减少IFN-γ的产生来增强Th2反应[34]。
4 AD炎症破坏角质形成细胞功能
已知FLG的功能缺失突变是AD最强的遗传危险因素,但研究显示没有FLG突变的AD患者皮肤中FLG表达减少也很明显[35]。除此之外,Cole等[36]发现没有携带FLG突变的AD儿童也存在脂质代谢过程的失调。所以说,除了遗传因素,还有其他机制参与皮肤屏障破坏的发生发展,即获得性皮肤屏障功能障碍。
在AD中,由Th2细胞、ILC2等2型炎症细胞分泌的炎症因子包括IL-4、IL-5、IL-13、IL-31等。KC表达IL-4和IL-13的受体复合物IL-4Rα/IL-13Rα1[37],且IL-4和IL-13均可结合IL-4Rα/IL-13Rα1并激活下游Janus激酶(Janus kinase,JAK)1/酪氨酸激酶(tyrosine kinase,TYK)2/JAK2,然后激活信号转导和转录激活因子(signal transducer and activator of transcription,STAT)6/STAT3。研究证明,AD患者皮肤KC和真皮神经纤维中可检测到IL-31的受体IL-31RA[38]。因此,2型炎症因子可通过直接作用于KC上相应的受体,成为AD皮损持续和加重的促成因素之一。
研究证明,2型炎症因子可通过改变KC间脂质成分、减少屏障蛋白等多种途径进一步破坏AD患者的皮肤屏障功能[39]。研究发现IL-4和IL-13是导致AD中FLG表达减少的原因之一[15]。IL-31可减少FLG、K10和IVL的表达[40]。此外,2型炎症因子还可促进短链脂肪酸和短链神经酰胺的产生[41],加剧表皮屏障缺陷。IL-4和IL-13还可刺激KC表达TSLP,进一步联系了皮肤屏障缺陷与Th2极化[42]。综上所述,2型炎症与获得性皮肤屏障破坏之间的确存在相互联系。
随后的研究进一步阐明了2型炎症导致KC功能异常的具体作用机制。Kim等[16]首次证明通过STAT6依赖机制,IL-4和IL-13可下调AD皮肤中LOR和IVL的表达。还有学者提出,IL-13/IL-4-受体-JAK激酶轴的激活可下调FLG、LOR和IVL的表达[37]。Berdyshev等[41]也发现IL-4和IL-13以STAT6依赖的方式通过抑制超长链脂肪酸延伸酶(elongation of very long chain fatty acids, ELOVL)3和ELOVL6造成皮肤屏障的破坏。总的来说,KC与2型炎症之间相互作用的恶性循环可导致AD的发生发展。
5 靶向创新药物对AD皮肤屏障的修复作用
近年来,许多靶向创新药物已应用于中重度AD的临床治疗,无论是生物制剂还是小分子药,对于AD的抗炎和止痒作用已得到临床验证。不少临床研究显示,AD患者应用新型靶向药物之后,皮肤屏障功能得到改善,屏障蛋白的含量增加[43-51](表1)。

表1 靶向创新药物对AD患者的抗炎及皮肤屏障修复作用的既往报道总结Table 1 Summary of previous reports on the anti-inflammatory and skin barrier repair effects of targeted innovative drugs on AD
度普利尤单抗(Dupilumab)是一种双靶点人源化单克隆抗体,通过特异性结合IL-4Rα亚基抑制IL-4/IL-13的信号传导。已有多篇报道证明了Dupilumab在AD中的抗炎和皮肤屏障修复作用[43-49]。JAK-STAT通路由配体或细胞因子与各种受体,包括JAK1、JAK2、JAK3和TYK2结合后的信号级联反应组成,参与多种AD相关细胞因子的信号传导,介导下游炎症反应[50]。Amano等[51]通过AD样皮炎小鼠模型证明JAK抑制剂可直接抑制KC中STAT3和STAT6的激活,进而改善皮肤屏障功能。临床研究方面,JADE MOA(NCT03915496)是一项应用阿布昔替尼(Abrocitinib)治疗AD的临床试验,研究者分析了活检皮肤中关键生物标志物治疗前后的表达变化,其中,表皮增生(K16)和Th2免疫反应(CCL17、CCL18、CCL26)等标志物与治疗前的水平相比均有所下降(https://clinicaltrials.gov/study/NCT03915496)。
6 结语与展望
在AD 2型免疫为主导的微环境中,KC与2型炎症之间密切交互作用,造成疾病的发生、发展与复发。靶向创新药物在展示抗炎治疗的同时,可能同时有效修复皮肤屏障,是AD治疗的有力武器。相信随着对疾病发病机制的深入研究,更多创新型药物将有望在抗炎、修复屏障方面双管齐下,成为实现AD治疗与管理的重要手段。
