CRISPR/Cas9技术高效制备山羊SOCS2基因编辑胚胎
2024-02-01张晨俭李隐侠刘伟佳王慧利吴家顺曹少先
张晨俭,李隐侠,丁 强,刘伟佳,王慧利,何 南,吴家顺,曹少先*
(1.南京农业大学动物科技学院,南京 210095;2.江苏省农业科学院畜牧研究所,江苏省畜禽精准育种工程研 究中心,农业农村部种养结合重点实验室,南京 210014;3.山东农业大学动物科技学院,泰安 271017)
细胞因子信号传导抑制因子2(suppressor of cytokine signaling 2,SOCS2),属于细胞因子传导抑制因子家族成员,是生长激素的负调控因子[1-3]。Metcalf等[4]研究发现,SOCS2基因敲除小鼠6周龄体重显著增加,Starr等[5]研究表明,SOCS2能通过与GH竞争GHR的结合,负调控GH-IGF1通路抑制生长。山羊SOCS2基因包含3个外显子,编码198个氨基酸。SOCS2蛋白包括3个结构域,中心保守的SH2结构域、C末端SOCS盒和可变的N-端结构域[1]。SH2结构域识别受体(如GHR)或者配体的磷酸酪氨酸是SOCS2蛋白发挥功能的关键步骤[6-8],识别过程中SH2结构域中5个高度保守的氨基酸残基(Arg73、Ser75、Ser76、Thr83和Arg96)通过氢键形成磷酸酪氨酸结合口袋,将磷酸化的酪氨酸残基(如GHR的pY595)锁定[9];N-端结构域包含一个扩展的 SH2子结构域 (ESS),连接SH2结构域和SOCS盒,使SH2结构域识别和捕获的底物泛素化[10]。据报道,绵羊SOCS2的SH2结构域第96号精氨酸突变为半胱氨酸(p.R96C),使其无法与磷酸酪氨酸结合导致SOCS2蛋白活性丧失,与野生型相比p.R96C突变绵羊体重增加了18%[11]。
CRISPR/Cas9属于第3代基因编辑技术,通过gRNA识别并引导核酸酶Cas9对靶基因进行切割产生双链断裂(DSB)[12],诱导DNA同源重组(HDR)[13-14]或非同源末端连接(NHEJ)修复[15],实现对靶基因编辑[16]。相较于前两代基因编辑技术ZFN、TALENs,CRISPR/Cas9具有编辑效率高、操作简便、成本低的优势,自问世以来,已被广泛应用于基因功能研究、疾病治疗和动植物育种等领域[17-19]。但CRISPR/Cas9的编辑效率和特异性与sgRNA序列直接相关[20-21],设计筛选特异性好、编辑效率高的sgRNA对于制备基因编辑动物特别是大动物至关重要。
目前关于山羊SOCS2基因编辑的报道很少,仅见Zhou等[22]利用碱基编辑器编辑绵羊SOCS2基因p.R96C的报道,且编辑效率仅25.0%(1/4)。亟需在山羊胚胎水平开展高效精准sgRNA的设计、筛选,为高效制备SOCS2基因敲除山羊,培育快长型肉用山羊提供技术基础。
1 材料与方法
1.1 试验材料
106枚新鲜波杂山羊卵巢采集自江苏省海安市屠宰场,浸泡于37 ℃生理盐水中,通过保温瓶带回实验室,进行卵母细胞分离和培养。
1.2 主要试剂
MEGAshortscriptTMT7 Transcription Kit、mMESSAGE mMACHINETMT7 ULTRA Transcription Kit、MEGAclearTMKit购于 Thermo fisher公司;QIAquick Gel Extraction Kit 购自Qiagen公司;GenCrispr Cas9 Nuclease 购于GenScript公司;pMDTM19-T Vector Cloning Kit、T4 DNA Ligase购自 TaKaRa 公司;Discover-sc Single Cell WGA Kit、DNA marker、2×Rapid Taq Master Mix、Phanta HS Super-Fidelity DNA Polymerase购自南京诺唯赞生物科技股份有限公司。
1.3 SOCS2 基因的sgRNA设计及载体构建
1.3.1 sgRNA引物设计 根据山羊SOCS2基因(XM_018047625)SH2的保守序列,使用在线网站CRISPOR(http://crispor.tefor.net/)设计山羊SOCS2基因的sgRNA靶序列,选择Lindel评分[23]高的sgRNA,其长度为20 bp,PAM(protospacer adjacent motif)序列为-NGG。为方便sgRNA引物与载体连接,在sgRNA正义链5′端添加ACCG,反义链5′端添加AAAC,见表1。送北京擎科生物科技有限公司合成。
1.3.2 sgRNA表达载体构建 使用BsaⅠ核酸内切酶将pGL3-U6-sgRNA-PGK-puromycin质粒线性化后胶回收纯化。分别将合成的sgRNA引物用超纯水稀释为100 mol·L-1,每对各取上下游5 μL混合,98 ℃变性10 min,室温冷却,在T4 DNA Ligase作用下与线性化质粒16 ℃连接过夜。转化DH5α感受态细胞,挑取菌落进行PCR鉴定。上游引物为pGL3-sg-F:5′-CGATTAGTGAACGGATCTCGACG-3′,下游引物为sgRNA反义链,扩增体系为2×Rapid Taq Master Mix 12.5 μL,上、下游引物各1 μL,菌液1 μL,加去离子水至25 μL;扩增程序包括95 ℃预变性3 min;95 ℃变性15 s,52 ℃退火20 s,72 ℃延伸15 s,共35个循环;72 ℃延伸10 min。PCR产物经琼脂糖凝胶电泳鉴定后选取阳性克隆送北京擎科生物科技有限公司测序。
1.4 sgRNA与Cas9 mRNA的体外转录
分别以质粒pX330和pGL3-U6-sgRNA-PGK-puromycin为模板,PCR扩增制备Cas9和sgRNA体外转录模板,将T7启动子引入扩增引物(序列见表1)。使用Phanta HS Super-Fidelity DNA Polymerase高保真酶进行扩增。扩增程序为:98 ℃预变性3 min;98 ℃变性10 s,60 ℃退火30 s,72 ℃延伸(Cas9、sgRNA体外转录模板延伸时间分别为2.5 min、30 s),共35个循环;最后72 ℃延伸10 min。对扩增产物进行胶回收纯化作为体外转录模板。Cas9 mRNA和sgRNA分别使用mMESSAGE mMACHINETMT7 ULTRA Transcription Kit和MEGAshortscriptTMT7 Transcription Kit进行体外转录,MEGAclearTMKit纯化,通过Nano drop2000超微分光光度计检测其浓度,琼脂糖凝胶电泳检测其完整性。将体外转录的sgRNA与Cas9 mRNA混合,调整其终浓度分别至50 ng·μL-1和100 ng·μL-1,置-80 ℃保存。
1.5 sgRNA/Cas9 蛋白体外切割活性测定
以山羊基因组序列为模板,使用Primer Premier 5在基因编辑靶区域上、下游500 bp附近设计检测引物SOCS2-F、SOCS2-R,序列见表2。以山羊全基因组DNA为模板,参照“1.3.2”的方法,对基因编辑区域进行PCR扩增。PCR产物胶回收纯化后进行体外切割。体系为:Cas9 蛋白5 U,sgRNA 0.2 μg,底物DNA 1 μg,10×Cas9 Buffer 5 μL,补充RNase free ddH2O至50 μL,对照组未加入sgRNA。37 ℃反应3 h后进行琼脂糖凝胶电泳检测。
1.6 卵母细胞体外成熟
参照Cripso等[24]的方法进行卵母细胞体外成熟培养。将收集到的卵丘-卵母细胞复合体(COCs)置于成熟培养基中,每孔500 μL成熟培养液中放入100个COCs并覆盖石蜡油,在38.5 ℃、5% CO2、饱和湿度培养箱中体外成熟培养24 h。
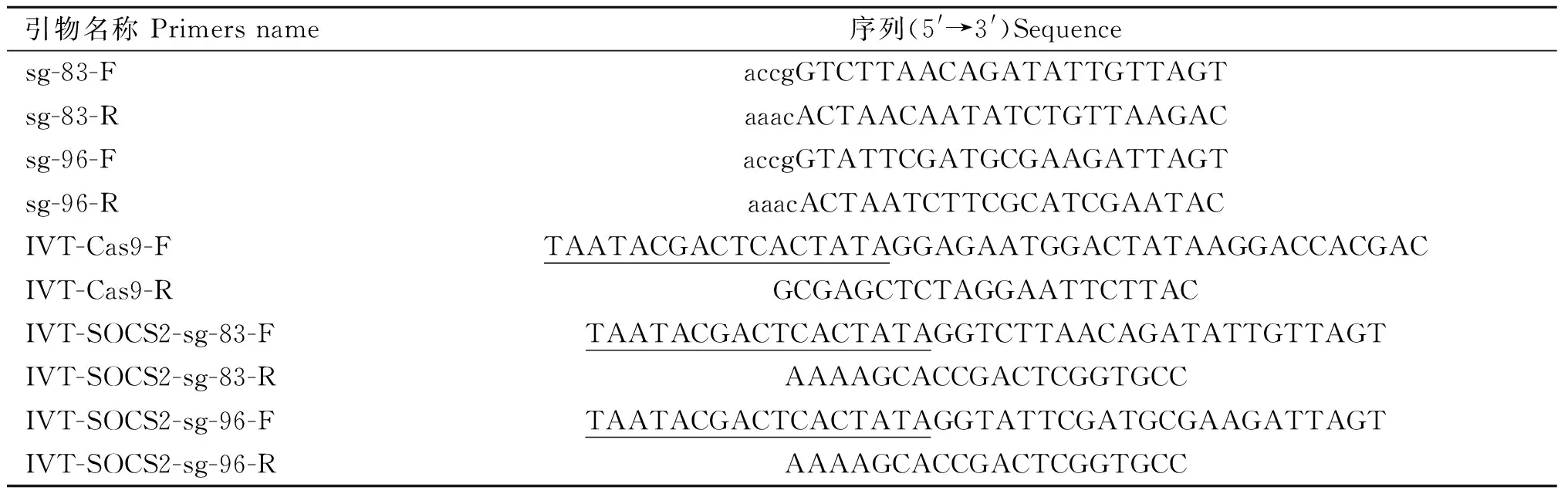
表1 sgRNA载体构建、Cas9和sgRNA体外转录模板制备引物Table 1 Oligonucleotides for constructing sgRNA vectors and making templates of Cas9 and sgRNA transcription in vitro
1.7 卵母细胞孤雌激活和显微注射
参照Ledda等[25]的方法对MⅡ期卵母细胞进行孤雌激活,激活后继续培养10~12 h,使用尼康显微注射系统对单细胞孤雌胚胎注射Cas9 mRNA和sgRNA的混合物,对照组注射无核酸酶的超纯水,继续培养 7 d。激活后48 h统计卵裂率,第7 天收集囊胚。
1.8 基因编辑效率的检测
收集单个囊胚于200 μL离心管中,按照Discover-sc Single Cell WGA Kit说明书进行单个胚胎全基因组DNA扩增。以扩增好的单个胚胎全基因组DNA 0.5~1 μL为模板,进行PCR扩增及测序鉴定,方法同“1.5”。在基因编辑靶区域附近出现双峰的测序样本,进一步进行克隆测序,每个样本随机选择10个阳性菌落,分析编辑形式及效率。
1.9 脱靶效应预测及检测
使用Cas-OFFinder(http://www.rgenome.net/cas-offinder/)在线网站对两条sgRNA靶序列进行脱靶预测。每条sgRNA选择5个错配数最少的潜在脱靶位点,设计OFF-Target 检测引物(见表2)进行PCR扩增测序,方法参照“1.3.2”。
2 结 果
2.1 山羊SOCS2基因sgRNA设计及载体构建
根据网站预测结果,如图1所示,选择SOCS2蛋白83号和96号氨基酸编码密码子附近的两条sgRNA,靶序列分别为,SOCS2-sg-83:TCTTAACAGATATTGTTAGT,SOCS2-sg-96:TATTCGATGCGAAGATTAGT。采用菌落PCR鉴定sgRNA表达载体,如图2所示,条带大小符合预期,Sanger测序表明插入序列及方向正确,载体构建成功。
2.2 Cas9 mRNA和sgRNA的体外转录
以pX330质粒和构建的sgRNA表达载体为模板,PCR制备Cas9和sgRNA体外转录模板,纯化后的产物如图3A、3B所示,条带单一,大小符合预期;以此模板进行体外转录的产物如图3C、3D所示,Cas9 mRNA和SOCS2-sg-83、SOCS2-sg-96主条带明显,片段大小符合预期,说明体外转录的mRNA质量较高。
2.3 sgRNA/Cas9蛋白体外切割活性检测
如图4A所示,对编辑区域进行PCR扩增,获得了条带单一,片段大小符合预期的切割底物,以此进行切割试验,结果如图4B所示,SOCS2-sg-83与SOCS2-sg-96两位点均切割完全,在250 bp和500 bp之间出现两条带,SOCS2-sg-96位点两条带大小比较接近,与预期SOCS2-sg-83位点切割后条带为287 bp和390 bp、SOCS2-sg-96位点切割后条带为319 bp和358 bp相符,表明两条sgRNA的体外切割效率高,可用于后续试验。
2.4 显微注射胚胎的体外发育
如表3所示,SOCS2-sg-83、SOCS2-sg-96分别注射胚胎152和140枚,48 h后卵裂率分别为75.7%和78.5%,囊胚率分别为55.7%和61.8%,与对照组无显著差异。说明本试验中的sgRNA和Cas9 mRNA未影响孤雌胚胎体外发育。

表2 基因编辑检测引物及潜在脱靶位点引物Table 2 Primers of gene editing detection and potential off-target sites detection

图1 山羊SOCS2基因sgRNA靶位点示意图Fig.1 Schematic diagram of the sgRNA target sites in the SOCS2 of goat

M. DNA相对分子质量标准;1~5、7~11. 阳性克隆;6. 阴性克隆 M. DL 2000 DNA marker; 1-5, 7-11. Positive colonies; 6. Negative colony图2 sgRNA表达载体PCR鉴定Fig.2 Identification of sgRNA expression vectors by PCR
2.5 CRISPR/Cas9编辑胚胎SOCS2基因效果检测
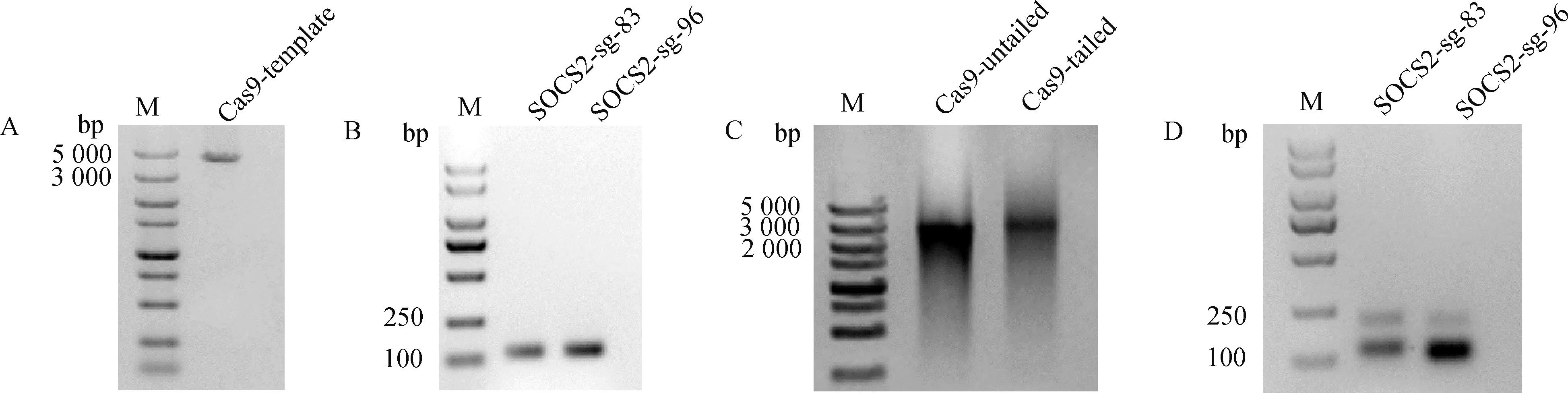
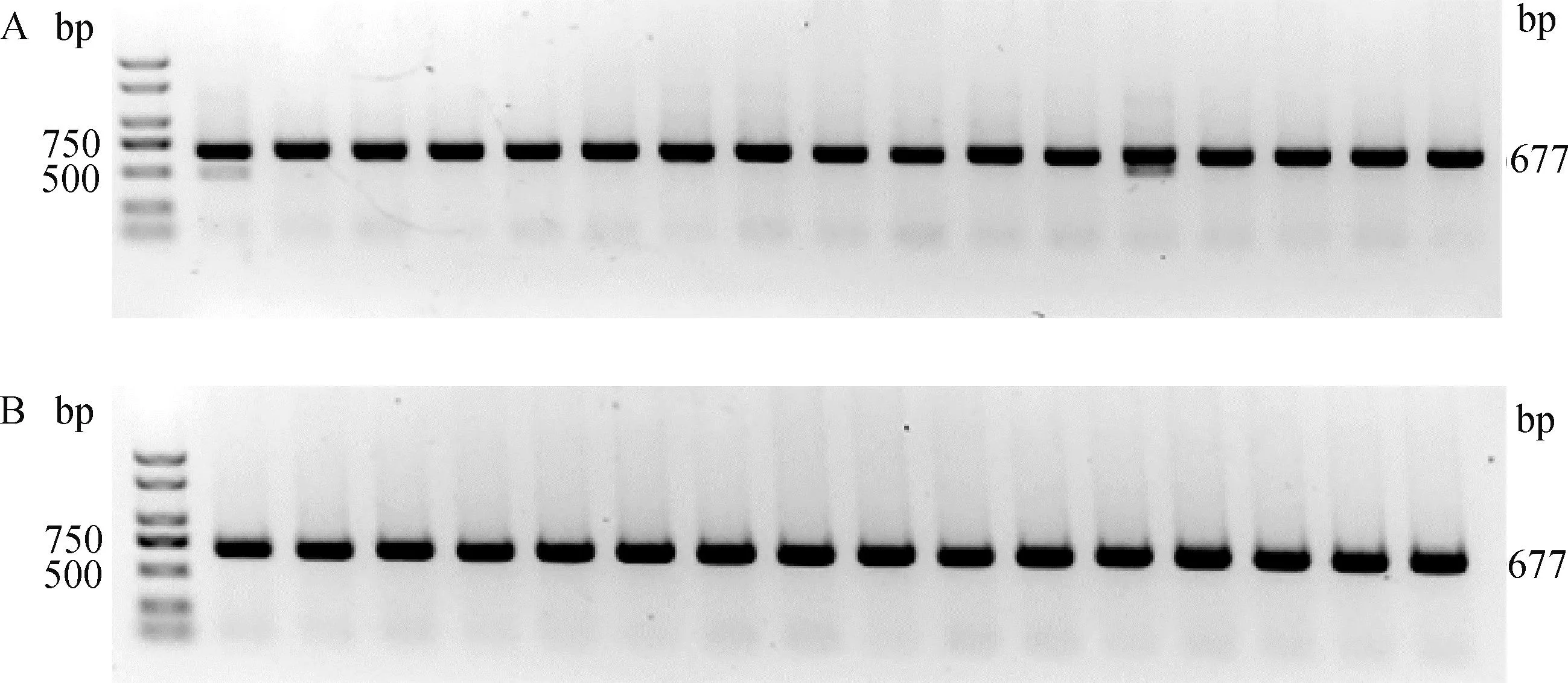
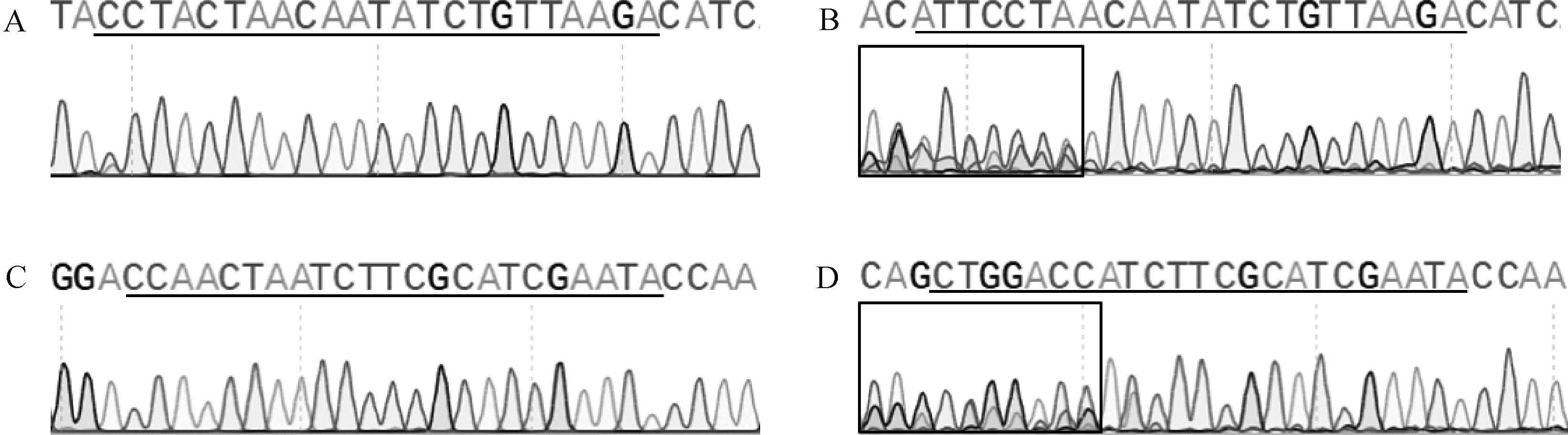
从64个注射SOCS2-sg-83的囊胚中随机选取17个进行全基因组扩增和PCR检测,如图5A所示,产物大小符合预期,83-1样本出现两条带,疑似出现大片段缺失。对PCR产物进行测序,其中有16个样本在PAM区附近出现双峰,如图6所示,说明在此处发生了基因编辑,效率为94.1%;从68个注射SOCS2-sg-96的囊胚中随机选取16个进行全基因组扩增和PCR检测,如图5B所示,产物条带单一,大小符合预期,测序后发现8个样本在PAM区附近出现双峰,效率为50.0%,见图6。同时,Cas9/sgRNA介导的突变发生在PAM区附近,符合Cas9切割的预期,进一步证明了该过程的靶向性。
A. Cas9体外转录模板制备:M. DNA相对分子质量标准;B. sgRNA 体外转录模板制备:M. DNA相对分子质量标准;C. Cas9 mRNA 体外转录:M. DNA相对分子质量标准;D. sgRNA 体外转录:M. DNA相对分子质量标准 A. Production transcription template of Cas9 in vitro: M. DL5000 DNA marker; B. Production transcription template of sgRNA in vitro: M. DL2000 DNA marker; C. Transcription of Cas9 mRNA in vitro: M. DL5000 DNA marker; D. Transcription of sgRNA in vitro: M. DL2000 DNA marker图3 Cas9和sgRNA体外转录Fig.3 Cas9 and sgRNA in vitro transcription

A. 切割底物PCR扩增;B. sgRNA/Cas9 体外切割活性测定:M. DL2000 DNA Marker;1. SOCS2-sg-83+Cas9;2. SOCS2-sg-96+Cas9;3. 对照组 A. Cleavage substrate PCR amplification; B. sgRNA/Cas9 cleavage activity detection in vitro: M. DL2000 DNA marker; 1. SOCS2-sg-83+Cas9;2. SOCS2-sg-96+Cas9;3. Control group图4 sgRNA/Cas9体外切割活性的检测Fig.4 Detection of sgRNA/Cas9 cleavage activity in vitro

表3 显微注射的山羊孤雌胚胎发育情况Table 3 Development rate of goat parthenogenetic embryos after microinjection
A. SOCS2-sg-83编辑囊胚PCR鉴定;B. SOCS2-sg-96编辑囊胚PCR鉴定 A. PCR identification of SOCS2-sg-83 edited blastocyst; B. PCR identification of SOCS2-sg-96 edited blastocyst图5 囊胚中CRISPR/Cas9编辑SOCS2基因效果的PCR检测Fig.5 PCR detection of the effect of CRISPR/Cas9 editing SOCS2 gene in blastocysts
A. 对照组SOCS2-sg-83靶位点附近峰图;B. 编辑组SOCS2-sg-83靶位点附近峰图;C. 对照组SOCS2-sg-96靶位点附近峰图;D. 编辑组SOCS2-sg-96靶位点附近峰图 A. Sequencing peak near SOCS2-sg-83 target site in the control group; B. Sequencing peak near SOCS2-sg-83 target site in the edited group; C. Sequencing peak near SOCS2-sg-96 target site in the control group; D. Sequencing peak near SOCS2-sg-96 target site in the edited group图6 囊胚中CRISPR/Cas9 编辑SOCS2基因效率的测序鉴定Fig.6 Sequencing identification of CRISPR/Cas9-edited SOCS2 gene efficiency in blastocysts
为了进一步研究CRISPR/Cas9在胚胎中的编辑形式,将PCR产物进行克隆测序,结果显示(表4),在SOCS2-sg-83编辑的160个克隆中152个出现插入、缺失或替换,概率为95.0%,最多的突变类型为插入一个T(概率为33.8%);移码突变(3N+1、3N+2)概率为81.3%(130/160)。此外,Indels为3N的克隆中有12个缺失83号氨基酸密码子,可能破坏SH2结构域;有两个插入了30个核苷酸,并引入了终止密码子(TAA);另一个插入了36个核苷酸,也引入终止密码子(TGA),将在翻译过程中提前终止。因此,共有90.6%的克隆可能实现对SOCS2基因功能的敲除。SOCS2-sg-96编辑的80个克隆中70个出现插入、缺失或替换,概率为87.5%,最多的突变类型为缺失4 bp,概率为36.3%(29/80),移码突变的概率为75.0%(60/80)。另外,在9个Indels为3N的克隆中,4个缺失第96号氨基酸密码子。共计80.0%(64/80)的Indels可能实现对SOCS2基因功能的敲除(表5)。
2.6 脱靶效应分析
根据Cas-OFFinder在线网站预测SOCS2-sg-83、SOCS2-sg-96的脱靶位点,结果显示,SOCS2-sg-83的预测位点中,错配数为2的有1个,错配数为3的有15个,错配数为4的有143个;SOCS2-sg-96的预测位点中,错配数为3的有2个;错配数为4的有25个。每个sgRNA选择5个错配数最少的脱靶位点进行PCR扩增测序,靶序列如表6所示,其中小写字母表示错配碱基。测序结果显示在选取的目标脱靶位点上未发现脱靶现象,如图7、图8所示。
3 讨 论
SOCS2是细胞因子传导抑制因子(SOCS)家族成员,通过其SH2结构域与GHR的磷酸化酪氨酸结合,阻断GH与GHR的结合,发挥抑制生长的作用[26]。组成SH2磷酸酪氨酸结合口袋的5个氨基酸残基(Arg73、Ser75、Ser76、Thr83和Arg96)在不同物种间高度保守,发生突变将影响其结合能力[9-10]。研究表明,SOCS2的p.R96C突变导致SH2与磷酸酪氨酸无法结合,SOCS2功能丧失[11]。本试验选择山羊SOCS2基因序列保守的83和96号氨基酸附近设计sgRNA,试图在基因编辑造成移码突变的同时也能通过破坏关键结构域导致基因的功能性敲除,提高基因敲除的效率,为高效制备SOCS2基因敲除山羊提供技术支撑。
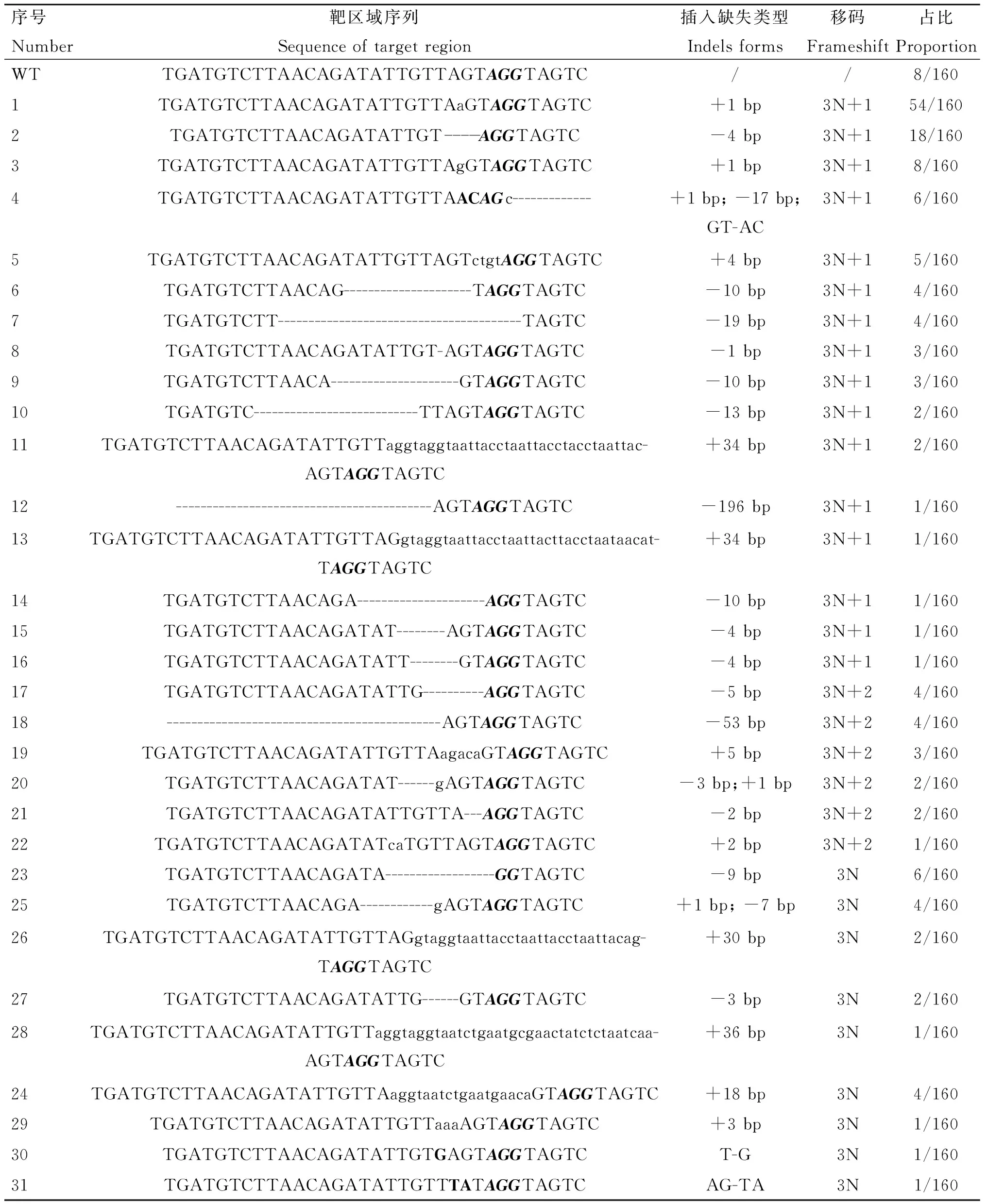
表4 SOCS2-sg-83不同编辑形式Table 4 Sequence varieties edited by SOCS2-sg-83
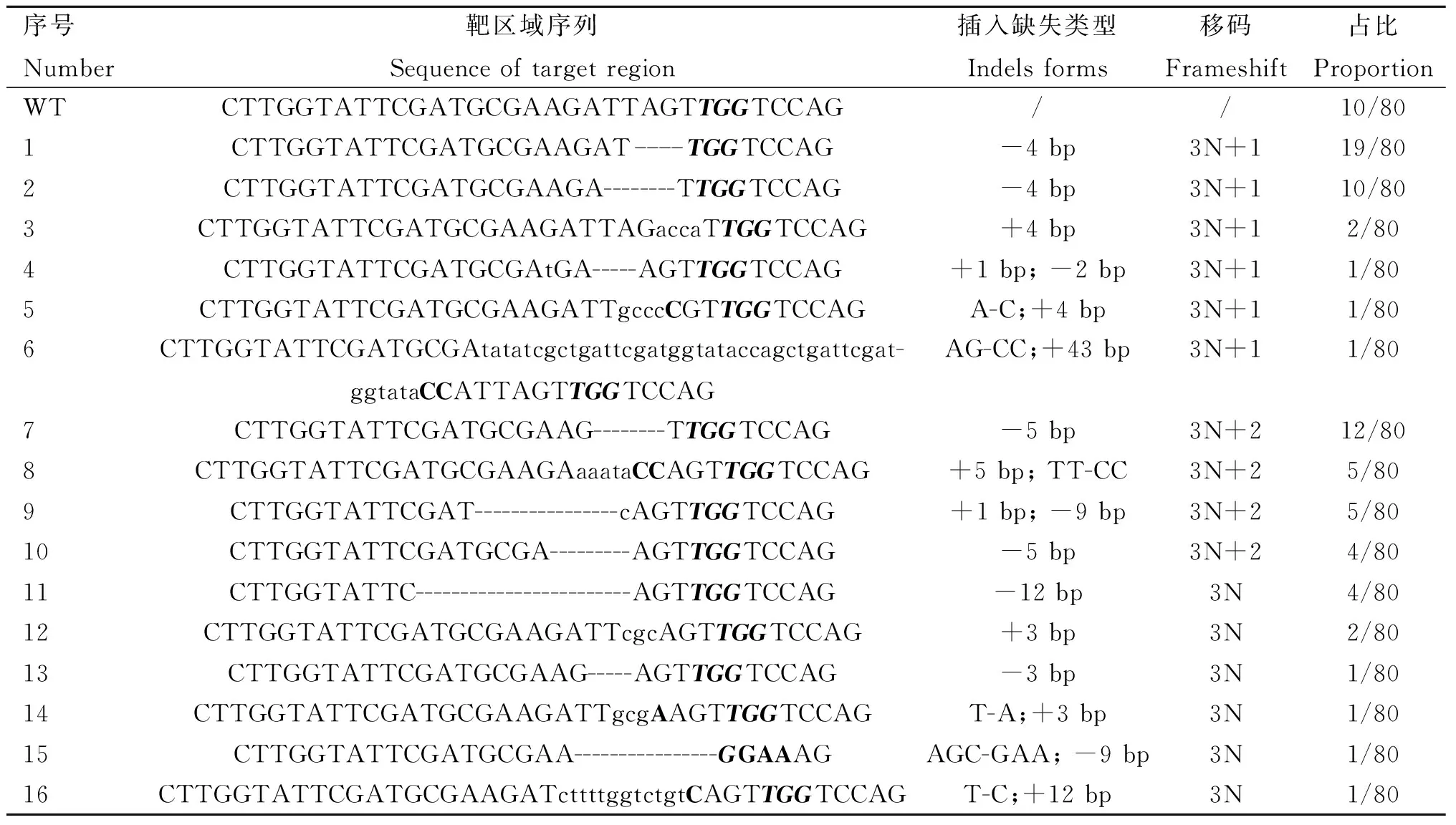
表5 SOCS2-sg-96不同编辑形式Table 5 Sequence varieties edited by SOCS2-sg-96
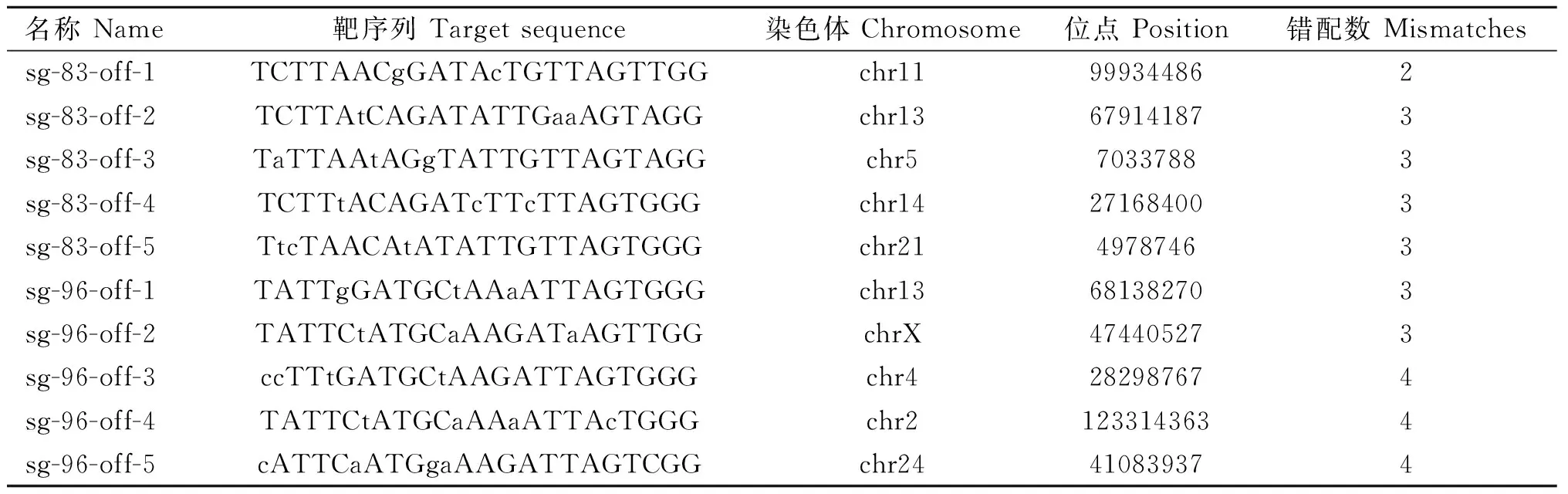
表6 sgRNA潜在脱靶位点预测Table 6 Potential off-target site prediction of sgRNA
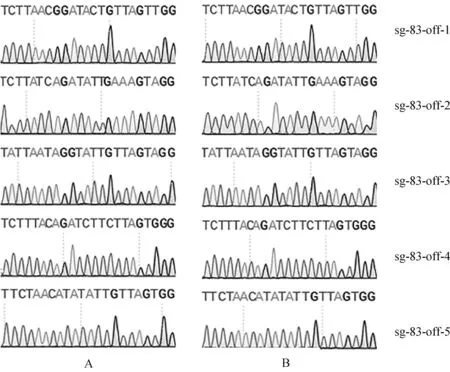
A.对照组;B.编辑组 A. Control group; B. Edited group图7 SOCS2-sg-83脱靶序列测序分析Fig.7 The off-target analysis of SOCS2-sg-83 by sequencing
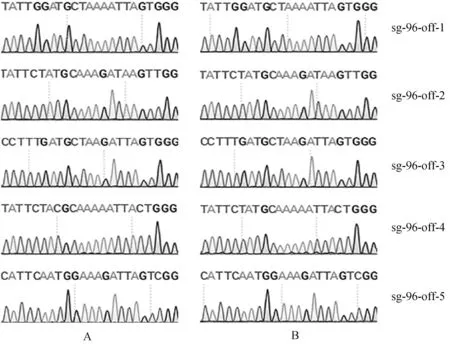
A. 对照组;B. 编辑组 A. Control group; B. Edited group图8 SOCS2-sg-96 脱靶序列测序分析Fig.8 The off-target analysis of SOCS2-sg-96 by sequencing
尽管CRISPR/Cas9系统的基因编辑效率相较ZFN、TALENs等技术大幅提高,但许多研究中基因编辑效率仍在50.0%以下。如尹智等[27]对猪孤雌囊胚α-1,3-半乳糖苷转移酶(GGTA1)基因的编辑效率为28.1%;尉翔栋等[28]编辑牛胚胎的肌肉生长抑素(MSTN)基因,编辑效率为15.4%;Zhang等[29]对绵羊孤雌胚胎的骨形态发生蛋白受体ⅠB(BMPR-ⅠB)基因进行编辑,效率为37.5%;目前,对SOCS2基因编辑的研究较少,仅见Zhou等[22]利用碱基编辑器对绵羊SOCS2的p.R96C位点进行编辑,效率为25.0%。这对于制备基因编辑动物是不利的,尤其是大型家畜的成本很高,有必要进一步提高编辑效率。研究表明,使用相同的基因编辑工具时,基因编辑的效率由sgRNA决定[30-32]。本试验通过设计、筛选获得的SOCS2-sg-83编辑胚胎的效率达到94.1%,可实现对胚胎的高效编辑。除了sgRNA的设计外,高质量转录的sgRNA和Cas9 mRNA也是本试验高编辑效率的重要保障。
基因编辑主要通过移码突变实现基因功能敲除[33-35],因为移码突变会破坏蛋白质的编码[36]。Zhang等[29]编辑绵羊胚胎BMPR-ⅠB基因,T-A克隆测序发现47.9%(78/163)的克隆存在移码突变。Hu等[37]对绵羊成纤维细胞生长因子(FGF5)基因进行编辑,造成缺失5 bp的移码突变,概率为66.7%(2/3),移码突变破坏了蛋白质一级结构并提前引入终止密码子,基因编辑羊的羊毛长度显著长于野生型(P<0.05)。本试验设计sgRNA时,选择了网站移码突变预测结果Lindel评分均为82的SOCS2-sg-83、SOCS2-sg-96,T-A克隆检测结果显示二者均可使SOCS2基因高效产生移码突变,其中SOCS2-sg-83引发移码突变的概率是81.3%,SOCS2-sg-96则为75.0%,高于66.7%的理论平均值。此外,在非移码突变的情况下,破坏关键结构域的核心氨基酸将导致蛋白质功能失活,从而实现基因功能敲除。如绵羊SOCS2的SH2结构域第96号氨基酸由精氨酸突变为半胱氨酸(p.R96C)可导致SOCS2功能失活,本试验中,SOCS2-sg-83引发的Indels为3 N的克隆中有7.5%(12/160)缺失83号氨基酸密码子,可能破坏SH2结构域;另外,有1.3%(2/160)插入了30个核苷酸,并引入了终止密码子(TAA),也可实现SOCS2基因功能性敲除。总之SOCS2-sg-83引发的突变中有90.6%可望实现SOCS2基因功能性敲除。此外,山羊SOCS2-sg-83在绵羊SOCS2基因SH2结构域中有完全匹配的靶序列,因此也可应用于绵羊SOCS2的基因编辑。
基因编辑中,sgRNA可能引导Cas9结合到与靶DNA相似的序列,产生非特异性编辑导致脱靶[38-40]。为此,本试验对每条sgRNA选择5个错配数最少的潜在脱靶位点进行了检测,未发现脱靶现象,说明SOCS2-sg-83、SOCS2-sg-96特异性较好,与郭日红[41]、赵为民等[42]的研究结果类似。此外,本试验中的sgRNA未影响孤雌胚胎发育,也从侧面印证了sgRNA的特异性。
4 结 论
本研究利用CRIPSR/Cas9 系统在胚胎水平获得了高效敲除山羊SOCS2基因的sg-SOCS2-sg-83,编辑效率高达94.1%,其中90.6%编辑可望造成SOCS2基因功能失活,且未发现脱靶,表明SOCS2-sg-83可实现山羊胚胎SOCS2基因的高效、精准编辑和敲除,为高效制备SOCS2基因敲除山羊,培育快长型肉用山羊奠定了技术基础。
