失活X染色体基因逃逸与系统性红斑狼疮的性别二态性
2024-01-24马茜周少岚党洁霍正浩马占兵
马茜,周少岚,党洁,霍正浩,马占兵
综 述
失活X染色体基因逃逸与系统性红斑狼疮的性别二态性
马茜1,2,周少岚3,党洁2,4,霍正浩2,4,马占兵2,4
1. 宁夏大学生命科学学院,银川 750021 2. 宁夏医科大学基础医学院医学遗传学与细胞生物学系,银川 750004 3. 宁夏医科大学总医院风湿免疫科,银川 750003 4. 宁夏医科大学教育部生育力保持重点实验室,银川 750004
X染色体失活可平衡女性中两条X染色体的基因剂量。越来越多的证据表明,失活X染色体上存在许多能够逃逸失活的基因。逃逸的机制涉及到DNA、RNA、组蛋白的表观修饰以及众多的调控蛋白和染色质的空间结构。失活X染色体基因逃逸的研究为人类疾病(特别是自身免疫性疾病)性别二态性的研究开辟了新的途径。目前已证实包括等失活X染色体基因逃逸是系统性红斑狼疮(systemic lupus erythematosus,SLE)女性好发的重要原因。本文主要综述了失活X染色体上基因逃逸以及与SLE性别二态性形成的分子机制。阐明SLE性别二态性形成的分子机制,不仅对疾病的诊断、治疗具有重要意义,而且对深入揭示人类免疫系统的发育及调控机理也有重要的理论意义。
失活X染色体;基因;逃逸;性别二态性;系统性红斑狼疮
系统性红斑狼疮(systemic lupus erythematosus,SLE)是一种临床常见的慢性、自身免疫性结缔组织病。由于T、B淋巴细胞异常活化,以及病理性自身抗体大量产生、补体激活、慢性炎症和免疫复合物沉积等病因,SLE患者往往出现皮肤、骨骼肌肉、肾脏、心肺及神经等多组织器官多系统受累的全身症状,但SLE确切的病因及发病机制仍不清楚[1]。
流行病学研究显示,我国SLE的发病率约为8.57/10万人[2]。SLE多发于育龄期青年女性,成年男女发病比例约为1∶9。此外,不同性别SLE患者的临床表现也具有明显的差异,男性SLE患者的临床表现发展更为迅速,器官损害更为严重,相较于女性患者,更容易出现心血管疾病、溶血性贫血、狼疮性肾炎、血管炎和神经精神性狼疮等并发症[3]。由此可见,不论在发病率还是临床表现上,SLE都呈现出与其他自身免疫性疾病相似的显著性别差异特征,即性别二态性(sexual dimorphism)[4,5]。
近年来,有关失活X染色体(inactive X chromosome,Xi)上的基因逃逸与SLE性别二态性的研究越来越受到关注。阐明自身免疫性疾病性别二态性形成生物学机制,不仅对疾病的诊断、治疗具有重要意义,而且对人类免疫系统的发育及调控也有重要的理论意义。本文综述了Xi基因逃逸的分子机制以及与SLE性别二态性的研究进展。
1 失活X染色体逃逸基因
人类()女性通常携带两条X染色体,男性携带一条X染色体和一条Y染色体。哺乳动物的性染色体起源于一对常染色体,经过大约1.7亿年的进化,由于突变、重复序列和缺失的累积过程,形成了现代人类性染色体模式,使X染色体携带的基因远多于Y染色体。目前发现,X染色体上有1669个基因,编码约800余种蛋白质,而Y染色体上只有426个基因,编码78种蛋白质,导致XX个体中X连锁基因的剂量大约是XY个体的两倍,对性别特异性发育及两性生存力的平衡极为不利。因而,在进化过程中,许多物种产生了伴性基因的剂量补偿机制(通过X染色体失活等途径)来平衡性别之间性染色体连锁基因的表达[6]。X染色体失活(X chromosome inactivation,XCI)是确保雌性(XX核型)和雄性(XY)细胞之间X连锁基因剂量补偿的表观遗传学机制,对于女性胚胎在发育过程中存活至关重要[7]。然而,并非所有Xi上的基因均处于沉默状态,总有一些基因能够从Xi逃逸,处于表达状态[8]。同时,Xi上逃逸基因的表达与自身免疫性疾病、癌症等人类疾病密切相关,为进一步揭示人类疾病性别二态性的发病机制开启了新的视角[9]。
1.1 逃逸基因的定位与类型
XCI理论提出之初并没有考虑到在X染色体上还有部分与Y染色体同源的基因,因此,Lyon[10]在提出X染色体失活假说后不久,就注意到X染色体上这些区域的基因可能存在失活逃逸,认为“如果X和Y的任何区域带有同源基因,则该区域很可能不会失活,因为不需要剂量补偿”。随后在人类和小鼠()中均证实,失活X染色体上的一些基因的确能够逃逸失活而表达[11],例如位于人Xp22.3拟常染色体区(pseudoautosomal regions,PARs)或附近的、、、等基因[12]。之后,人们又发现,Xi 逃逸基因并不仅仅限于PARs,例如编码锌指蛋白的基因位于Xp22.1和编码核糖体蛋白S4的基因位于Xq13[12]。Carrel等[13]发现,Xi逃逸的基因有21%位于X染色体短臂,只有3%位于长臂,在健康女性中有超过15%~30%的X连锁基因逃逸失活。在人类中,大约15%的基因始终能无选择的从失活中逃逸,被称为组成性逃逸(constitutive escape),另外15%的失活逃逸基因具有个体或组织之间的差异,被称为可变性逃逸(variable escape)[14,15]。迄今为止,已发现了100多个Xi逃逸基因,与自身免疫性疾病、癌症、神经系统疾病等许多人类疾病有关,成为人类疾病性别二态性研究的热点问题[16]。
1.2 逃逸基因的异质性
X染色体失活是伴随真兽类和有袋类哺乳动物性染色体演化而发生的普遍的生物学现象,在目前已研究的有限物种中,并非所有X连锁基因都被失活X染色体沉默,由此来看,Xi上的逃逸基因同样存在于不同的物种之间。Al-Nadaf等[17]通过RNA- FISH技术,分析了非洲象()、小鼠和人成纤维细胞系中的XCI,发现这些物种中均可观察到失活X染色体基因逃逸现象,只是人和非洲象的成纤维细胞中,逃逸失活的基因明显多于小鼠。有袋动物XCI中有14%~30%的基因逃逸失活,小鼠X连锁基因中,只有3%~7%始终逃逸XCI。另外一些研究也证实,失活X染色体逃逸基因具有物种间的差异[9,18~20]。
人类Xi逃逸基因的研究已历经了近40年的历史,2015年,Bradley等[21]对前期利用不同研究方法、大规模的研究结果进行了比较分析,尽管不同的研究方法所得结果不尽相同,但所有的研究都证实,Xi逃逸基因具有明显的异质性。2016年,Zhang等[22]采用二代测序技术,分析了从具有北欧和西欧血统的美国人和尼日利亚约鲁巴人B淋巴细胞系获得的RNA和基因型测序数据,鉴定出114个X染色体失活逃逸基因,这些基因的分布既有种族间的差异,又有个体间的差异。
2017年,Tukiainen等[23]对染色体失活逃逸基因进行了系统分析,结合基因组序列数据,整合了来自GTEx和940个单细胞转录组的29个组织、449个个体5500多个转录组的数据,从群体水平、个体水平和单细胞水平对Xi逃逸基因进行了系统研究。结果表明,Xi上有82个组成性逃逸基因,89个可变逃逸基因和390个失活基因,逃逸基因有显著的性别差异;在Xq区域,许多逃逸基因呈现出高度的组织特异性,其中基因只在肺组织中逃逸,基因在不同性别中呈现出组织差异性。随后,Garieri等[24]将深度单细胞RNA测序与全基因组测序相结合,分析了5个女性个体的935个原代成纤维细胞和 48 个淋巴母细胞单细胞中的等位基因特异性表达,鉴定出 55 个逃逸基因。所有基因在每种细胞和细胞类型中均表现出可变的逃逸倾向,并且每个细胞均显示出逃逸基因的独特表达谱。
最近,Zito等[25]分析了来自twins UK bioresource数据集中248对女性双生子个体的脂肪组织、皮肤组织、淋巴母细胞样细胞系和纯化的免疫细胞(包括:单核细胞、B细胞、T-CD4+、T-CD8+和NK细胞)Xi逃逸基因的分布特征,确定了62个逃逸基因,包括19个lncRNAs基因。发现单卵双生子比双卵双生子具有更多的逃逸基因,表明遗传因素可能是个体间逃逸基因差异的基础;同时,在单卵双生子中,逃逸基因也存在着一定的差异,表明环境因素也可影响失活基因的逃逸。不同组织中的逃逸发生率不同,脂肪组织(26%)和皮肤组织(29%)高于淋巴母细胞样细胞系(16%);在不同的免疫细胞中,基因逃逸的发生率也有显著的差异,单核细胞为15%、B细胞为20%、T-CD4+为22%、T-CD8+为25%、NK细胞为29%,在不同类型的免疫细胞中逃逸基因谱系具有明显的异质性。X染色体富含具有免疫和神经调节功能的基因[26,27],因此,逃逸基因的异质性,可能是女性表型和疾病风险性别二态性的重要原因[28~30]。
1.3 基因逃逸的分子机制
XCI的发生与维持是一个非常复杂的过程,包括计数、选择、启动、扩散及维持,伴随DNA甲基化、组蛋白修饰、m6A修饰、多种相关蛋白复合物募集、染色质空间结构改变等一系生物学变化,需要对许多不同因素进行准确的时空调节,以实现全染色体范围的基因沉默[31,32]。Xi上的基因如何实现由沉默到表达的逆转,依然是非常复杂的过程,同样涉及到DNA、RNA、组蛋白的表观修饰,涉及到众多的调控蛋白以及染色质的空间结构。
1.3.1 组蛋白修饰
组蛋白翻译后的甲基化和乙酰化修饰,可通过影响染色质的空间结构以及启动子、转录区、增强子和绝缘子等功能元件,分割染色质不同的功能区域,调控基因沉默与表达。处于不同功能状态的基因,组蛋白修饰的谱系不同。在转录活性基因的启动子区,通常有H3K4me2、H3K4me3、乙酰化和H2A.Z标记;转录区域富集H3K36me3和H3K79me2,而H3K9me2和/或H3K9me3、H3K27me3往往富集在处于沉默状态基因的特定结构域中[33](图1)。
许多研究表明,XCI的启动与维持以及Xi上基因的逃逸与组蛋白修饰密切相关。Xi上处于沉默状态的基因与逃逸基因具有不同的组蛋白修饰标记[9]。沉默基因常位于Xi具有典型抑制标记区域,例如H3组蛋白第9、20、27赖氨酸残基的三甲基化(H3K9me3、H4K20me3、H3K27me3)区域[34],H3赖氨酸残基的甲基化修饰能够募集多梳复合物,调节染色质结构,使染色质处于紧密状态,从而抑制基因的转录活性[35]。而逃逸基因则常位于Xi具有典型活性标记区域,如H3K4me2、H3K4me3、H3K9me1以及H3K9ac、H3K27ac[36,37]。还发现沉默基因区域有lncRNA和果蝇基因增强子的人类同源物2 (enhancer of zeste homolog 2,EZH2)富集。Xist RNA与X染色体的结合是失活的关键一步,EZH2是多梳抑制复合体2 (polycomb repressive complex 2,PRC2)的催化亚基,可通过组蛋白甲基转移酶 (histone methyltransferase,HMTase)催化组蛋白H3 第27位赖氨酸三甲基化,沉默靶基因转录[38,39],而在逃逸基因位置这些抑制因子被耗竭[40]。
有研究表明,H3K4甲基化和H3/H4乙酰化在Xi逃逸基因的启动子区存在共定位。Brinkman等[41]通过ChIP实验发现,H3/H4乙酰化和H3K4me3共定位在Xi逃逸基因磷酸化酶激酶调节亚基α1 (phosphorylase kinase regulatory subunit alpha 1,)的活性启动子区域,转录起始位点区域这种共定位修饰的存在与基因活性呈正相关,表明组蛋白修饰在Xi基因逃逸过程中具有重要作用。
1.3.2 DNA重复序列
DNA重复序列在基因重组中扮演着重要的角色,是染色质高级结构组织的驱动力,可驱动染色质的定点折叠和染色质交联,在Xi的形成及基因逃逸失活的过程中具有重要作用[42,43]。1998年,Lyon[44]推测X染色体上长分散重复元件1 (long interspersed repetitive elements,LINE1)可能是兼性异染色质形成的原因。后续的许多研究证实,人类X染色体的LINE-1分布与常染色体不同,LINE-1富集在X染色体上,是常染色体的2倍;Xi 上LINE-1主要富集于失活基因周围,而在逃逸基因周围较少,表明LINE1有助于Xist RNA锚定在X染色体上,促进Xist RNA沿X染色体扩散,使X染色体上的基因发生沉默[45]。Chow等[46]报道,LINE1参与了Xist RNA诱导的异染色质核区室的组装,沉默的核区室缺失RNA聚合酶II (Pol II)和转录因子。在小鼠胚胎干细胞的分化过程中,沉默基因最初位于Xist RNA区室的外部,随着XCI的进行,基因进入Xist RNA区室,进而失活;而从Xi逃逸的基因则留在外部,表明逃逸基因和失活基因表现出不同的核组织,提示LINE-1可能在这种空间分离中起作用[47]。
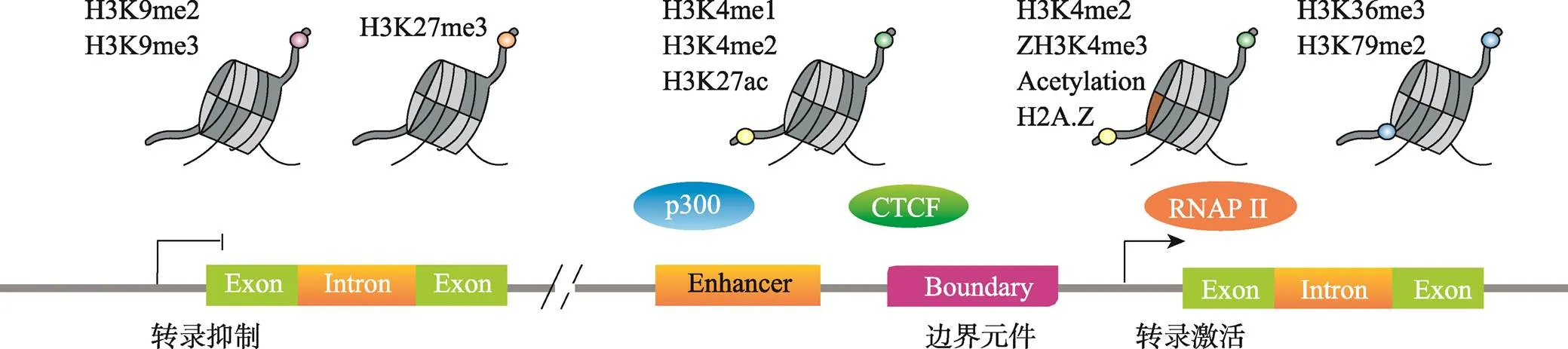
图1 哺乳动物基因组中不同功能元件的组蛋白修饰
活性启动子通常由组蛋白H3K4me2、H3K4me3、乙酰化(ac)和H2A.Z标记。转录区域富集H3K36me3和H3K79me2。抑制基因位于H3K9me2和/或H3K9me3或H3K27me3的结构域中。增强子相对富集H3K4me1、H3K4me2、H3K27ac和组蛋白乙酰转移酶p300。CTCF结合许多可能起边界元件、绝缘体或结构支架作用的位点。染色质的这些不同特征有助于组织DNA并区分基因组中的功能元件。根据文献[33]修改绘制。
除LINE-1元件外,其他重复序列也可能在基因逃逸中发挥作用。2006年,Wang等[43]报道,人类大多数Xi上失活基因富含AT短重复序列,相反,逃逸基因缺少AT短重复序列,表明AT基序参与了Xi上基因逃逸的过程。Nguyen等[48]报道,小鼠Xi上AT基序的富集程度比人类Xi更高,或许这与小鼠X染色体失活逃逸基因比人类更少有关;不论是小鼠还是人类,逃逸基因中AT基序富集程度均低于失活基因。AT基序可招募AT富集序列结合蛋白1 (special AT-rich sequence-binding protein 1,SATB1)和核异质核糖核蛋白U (heterogeneous nuclear ribonucleoprotein U,HNRNPU),参与染色质构象的变化[49,50]。SATB1与核基质中的碱基非配对区结合,使染色质折叠形成环状结构[51];还可通过调节组蛋白乙酰化和甲基化等多种途径调控基因表达[52]。病毒SATB1的表达可使小鼠胚胎成纤维细胞中的Xist基因沉默,相反肿瘤细胞中SATB1的缺失,能使Xist的沉默功能丧失[50];HNRNPU也被称为核支架附着因子(scaffold attachment factor A,SAF-A),是hnRNPs 家族中的一员,属于核基质蛋白,HNRNPU 与Xist RNA相互作用,介导RNA Xist结合到X染色体上并招募更多的沉默复合物,从而使X染色体失活[53]。
1.3.3 染色质空间结构
染色质空间结构的变化也是导致基因逃逸的重要因素。目前的研究表明,CCCTC结合因子(CCCTC-binding factor)作为一种关键的染色质架构蛋白,可与黏连蛋白(cohesin)结合,在染色质三维结构的形成以及Xi基因逃逸中发挥着重要作用。
CTCF是一种在进化上高度保守的锌指蛋白,能与特定基因结合发挥多种功能[47,54]。2005年,Filippova等[55]报道,与活性X染色体相比,Xi上结合的CTCF明显减少,有趣的是,Xi上结合的CTCF主要富集于逃逸区;在小鼠中,CTCF与和两个组成性逃逸基因的5′端结合,形成Xi上沉默区域和逃逸区域之间的边界,起到绝缘子的作用,阻断异染色质传播。最近,Fang等[56]系统研究了Patski细胞和成年小鼠F1杂交组织中组成性和可变逃逸等位基因特异性CTCF结合模式和表观遗传学特征。研究表明,当Patski细胞中Xi上CTCF边界区P4被删除时,逃逸完全丧失,但在大脑中基因并未逃逸,同时发现,在大脑中周围没有CTCF结合。因此,CTCF可能主要参与染色质结构的隔离;相反,YY1在调控启动子-增强子相互作用中发挥直接作用,并在逃逸基因的TSS处富集;作者认为,与组成性逃逸基因相关的调控元件可能与兼性逃逸基因不同,导致对边界缺失的不同影响,或许这可以部分解释失活基因逃逸的组织特异性[57,58]。另外,Sun等[59]报道,CTCF也是一种RNA结合蛋白,在XCI前期,CTCF蛋白能抑制的转录;在XCI开始时,Jpx RNA表达上调并与CTCF结合,去除CTCF的抑制作用,激活Xist表达,表明CTCF在XCI和失活基因逃逸过程中似乎扮演着多种角色[60]。
总之,失活X染色体基因逃逸的分子机制研究已经取得了长足的发展(图2)。但由于这个过程涉及到DNA甲基化、组蛋白修饰、m6A修饰、多种非编码RNA的调控、众多相关蛋白复合物募集以及染色质空间结构改变等一系生物学过程,同时还表现为明显的个体、组织以及细胞间异质性,相关分子机制依然还有许多未解之谜。
2 失活X染色体基因逃逸与SLE
长期以来,人们一直认为Xi基因逃逸会导致疾病的性别差异。由于X染色体携带着高密度的免疫相关基因,因此,Xi基因逃逸可能导致女性位于X染色体的免疫基因剂量高于男性,形成自身免疫性疾病的性别差异[61,62]。目前,已确定有大约20多个免疫相关基因能从Xi逃逸,约占X染色体所有表达基因的10%[63~65],其中一些基因对SLE性别二态性的形成具有重要作用(表1)。
2.1 TLR7
(Toll-like receptor 7,TLR7)基因位于Xp22.2,在浆细胞样树突状细胞、单核细胞/巨噬细胞和B细胞中表达。基因产物是一种细胞内受体,位于内体和溶酶体中,可与单链RNA (ssRNA)及核糖体中ssRNA降解的产物-鸟苷和寡核苷酸结合,这种双重结合可能协同增强的激活,激活后的具有广泛的免疫调节功能。在DC细胞中,TLR7刺激产生B细胞激活因子,调节B细胞增殖、分化和存活,增强自身反应性B细胞的激活和自身抗体的产生[66,67]。异常激活可促进SLE的疾病进展[68];TLR7还能刺激TNF-a、IL-6、IL-12等促炎细胞因子产生,并促进Th1极化;通过干扰素调节因子5、NF-κB和AP-1等转录因子介导调节趋化因子配体5 (chemokine ligand 5,CCL5)分泌;有研究表明,TLR7能通过干扰素调节因子7 (interferon regulatory factor 7,IRF7)触发I型干扰素的分泌,I型干扰素水平升高和干扰素信号基因表达增加是SLE最显著的特征[64,69]。
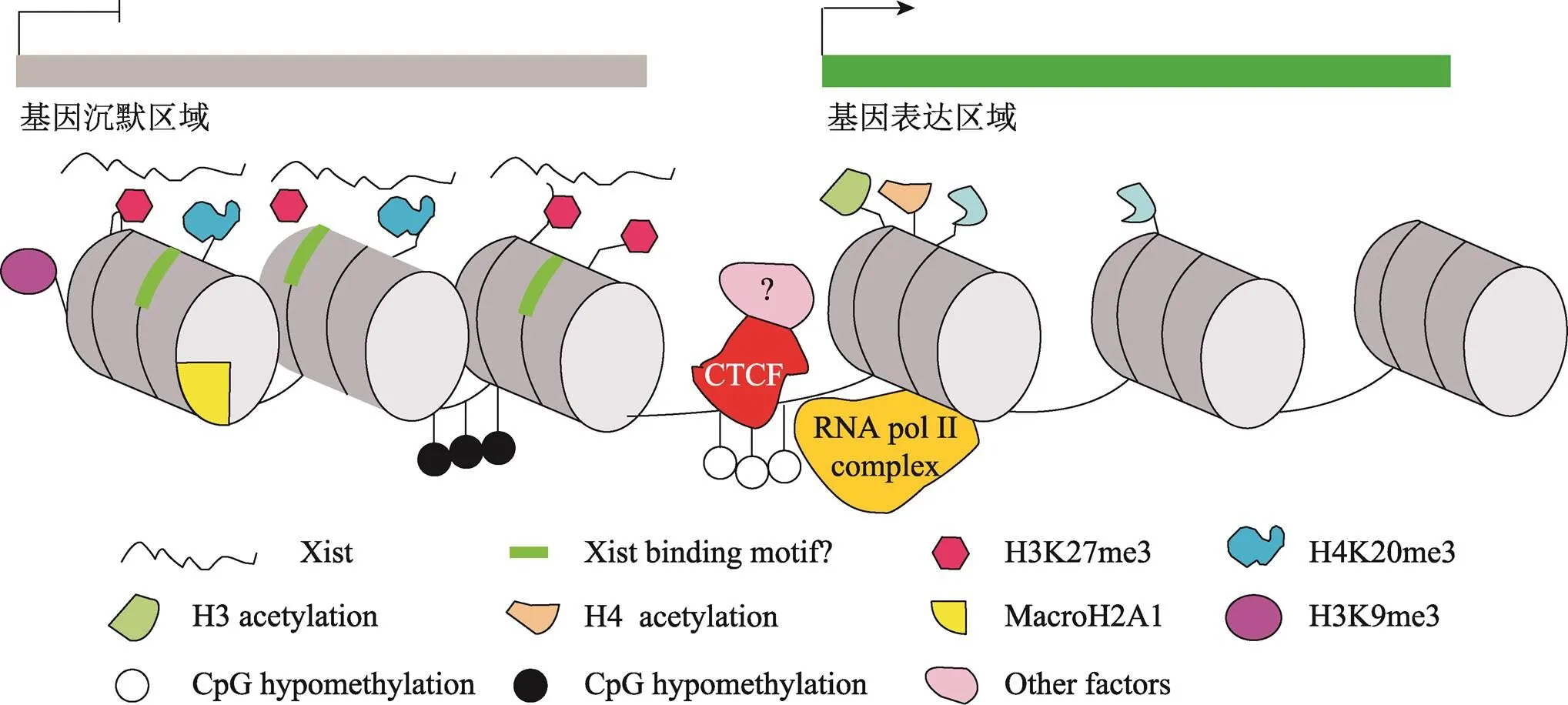
图2 失活X染色体基因逃逸的分子机制
Xi中沉默的染色质被Xist RNA包裹,可能通过特定的DNA基序。抑制性组蛋白修饰和组蛋白变体(例如,H3K27me3、H3K9me3、H4K20me3和macroH2A1)被募集,DNA甲基化修饰CpG岛。这种类型的染色质结构阻止转录。相反,逃逸基因区域富含经典的活性组蛋白标记(例如,H3K4me3、H3和H4乙酰化)和RNA聚合酶II (RNA pol II),并且在其CpG岛处被低甲基化。由绝缘体蛋白CTCF结合的绝缘体位点,可能将失活基因与活性基因分离。CTCF结合可以阻断CpG甲基化和抑制性染色质的扩散。根据文献[8]修改绘制。
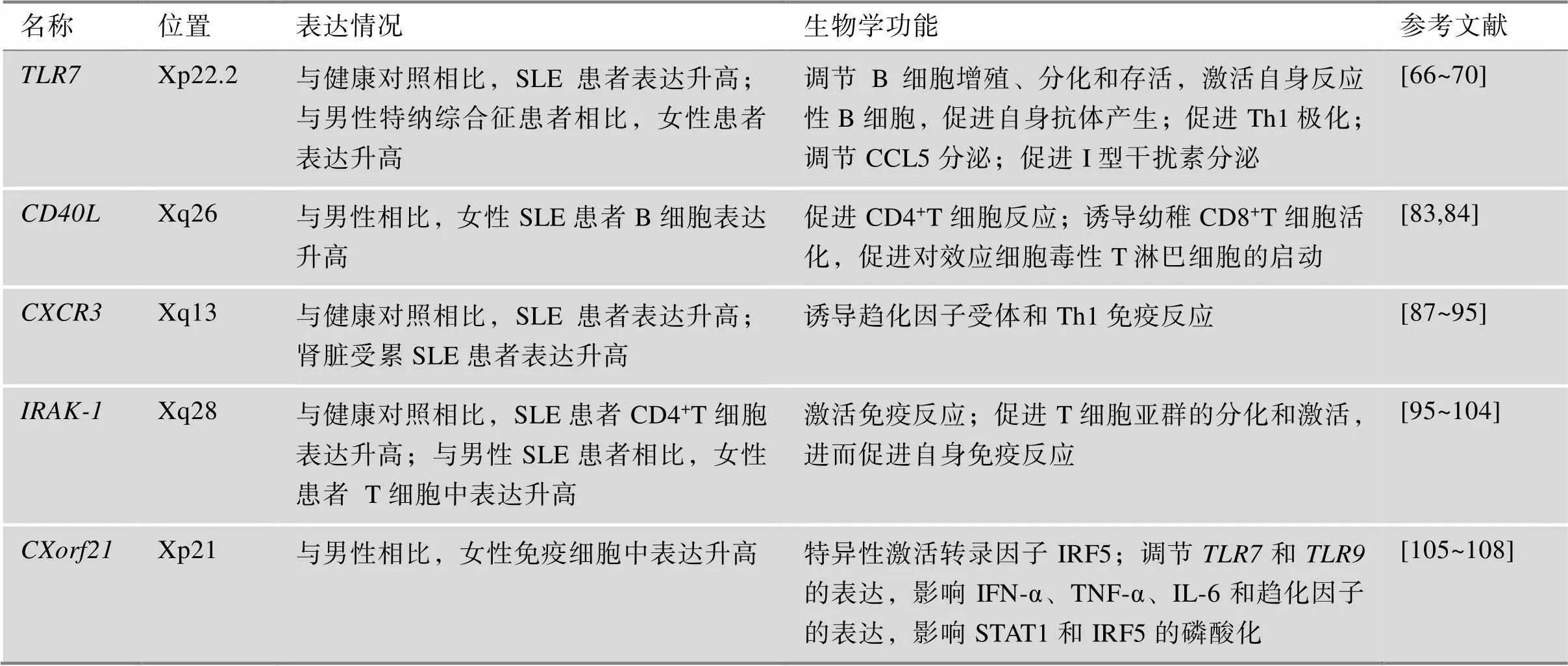
表1 与SLE性别二态性相关的Xi逃逸基因
早期的一些研究表明,雄性BXSB小鼠比雌性小鼠SLE的发病率要高得多,并且发病较早。 BXSB是由雌性C57BL/6(B6)小鼠和雄性SB/Le小鼠杂交形成,携带有Y连锁自身免疫加速器(Y chromosome- linked gene for accelerated autoimmunity,Yaa)片段,该片段属于拟常染色质区域,长4 Mb[70~72]。Amano等[73]发现,带有Yaa片段的B细胞表现出自发性IgM分泌增加,同时,缺乏边缘区B细胞(marginal zone B cells,MZB),表明带有Yaa片段Y的B细胞过度活跃。之后,Pisitkun等[74]鉴定了Yaa基因产物,并分析了Yaa如何改变自身抗体对核仁抗原的特异性。通过对滤泡B细胞RNA的微阵列分析,比较了B6Yaa小鼠和B6野生型小鼠中的基因表达,发现B6Yaa小鼠的B细胞中有26个基因显著上调,其中位于X染色体远端的和表达增加了一倍,定量PCR和蛋白质印迹分析证实,Yaa中的基因组重复导致B细胞中TLR7 mRNA和蛋白质的增加;在携带Yaa Y染色体的BXSB小鼠中也证实了基因的重复;同时,发现可能通过诱导含有核仁来源的RNA抗原,激活B细胞,促进自身免疫。
在人类中,X染色体数目异常患者已成为研究X染色体失活逃逸基因的重要病理模型。许多研究已证实,随著X染色体的数目的增加,SLE等自身免疫性疾病的发病率也随之增加[65,75]。例如:Klinefelter综合征(47,XXY)SLE的发病风险比正常男性(46,XY)增加了14倍,47,XXX女性SLE的发病风险是正常女性(46,XX)的2.5倍,相反,Turner综合征(45,X)SLE发病风险比正常女性(46,XX) 低[76~79]。这些结果表明,X染色体促进了不依赖激素的自身免疫性疾病的发生,是自身免疫性疾病易感性的一个重要风险因素。临床研究表明,在SLE患者中,由于基因拷贝数增加而过度表达。与正常男性(46,XY)外周血单个核细胞相比,正常女性(46,XX)和Klinefelter综合征(47,XXY)患者的表达水平显著升高[66];Souyris等[69]报道,来自正常女性和Klinefelter综合征患者的幼稚B淋巴细胞、单核细胞和浆细胞样树突状细胞中存在双等位基因,表达水平显著增高,导致B细胞产生的IgG抗体是单等位基因B细胞的两倍多。这些研究结果证实,从失活X染色体逃逸的基因是SLE性别二态性形成的原因之一。
2.2 CD40L
(cluster of differentiation 40 ligand,CD40L)基因定位于Xq26,编码的CD40L是一种II型跨膜蛋白,属于TNF家族成员。CD40L由261个氨基酸组成,分子量为35 kDa,包含一个氨基端胞内结构域、一个跨膜结构域和一个羧基端胞外结构域,主要表达于活化的CD4+T细胞和血小板。CD40与CD4+T细胞上的CD40L及DC结合,增加主要组织相容性复合体(major histocompatibility complex,MHC)分子的表达以及共刺激信号,包括CD80、CD86和DC中的细胞因子,这些分子反过来会增加CD4+T细胞反应;激活的CD40L还能通过DC诱导幼稚CD8+T细胞活化,促进对效应细胞毒性T淋巴细胞的启动[64]。
以往的研究表明,CD40-CD40L通路是SLE发病机制和自身抗体产生及其在肾脏中沉积的核心,是导致狼疮肾炎患者肾损伤的重要因素。T细胞上的CD40L和肾间质中B细胞上的CD40之间的相互作用,介导髓样细胞和驻留肾细胞的激活,进一步放大间质和肾小球中的炎症环境[64]。Koshy等[80]报道,体外培养的SLE 患者T细胞中CD40L的表达量与健康对照相比显著上调,培养24小时后,CD40L的表达量持续上升至,并且在培养48小时后继续表达;活动性狼疮患者B细胞中CD40L的表达增加了20.5倍。的高表达不仅发生在T细胞中,而且发生在狼疮患者B细胞中[81]。问题是,的高表达到底是位于Xa上的基因过表达,还是Xi上的逃逸了失活?首先,临床研究发现,与健康男性、Turner综合征相比,Klinefelter综合征的表达水平显著增高,CD3+CD40L+T细胞数量增加,表明的高表达与X染色体的数目有关[66];其次,女性SLE患者CD4+T细胞过表达,其原因是启动子区域被去甲基化,表明不是从Xa过表达的,而是从重新激活的Xi表达的[82-84];再次,通过XIST RNA探针识别Xi,应用RNA FISH和DNA FIS检测和的表达,结果表明,与正常B细胞相比,SLE B细胞核内缺失RNA转录物积累的细胞更多,同时表达增高[85]。这些研究结果证实,从Xi逃逸的是女性SLE发病风险的重要因素之一,也是SLE性别二态性形成的原因之一。
2.3 CXCR3
(DEAD-box helicase 3 X-linked,DDX3X)定位于Xq13,编码一种G蛋白偶联受体,由368个氨基酸组成,分子量约为41 kDa。目前已发现3种可变异构体:CXCR3-A、CXCR3-B和CXCR3-ah,通常所说的CXCR3是指CXCR3-A[86,87]。表达于活化的CD4+T、CD8+T、巨噬细胞、树突状细胞等免疫细胞,可诱导趋化因子受体和Th1免疫反应[88]。在免疫细胞中,对表达的调控至关重要,以防止免疫反应的失控。
对模型动物研究表明,在狼疮小鼠CD4+T细胞中,的表达增加。在KO狼疮小鼠中,CD4+T细胞向B细胞滤泡的迁移和T辅助功能降低,自身抗体的产生下降,同时Tfh细胞、生发中心的B细胞和浆细胞的比例也降低,表明CXCR3可能通过增加异常激活的Tfh细胞和B细胞的百分比以及促进狼疮小鼠中CD4+T细胞的迁移和T辅助功能,在自身抗体产生中发挥重要作用[89]。在狼疮性肾炎的动物模型中,CXCR3及其配体CXCL9通过活化的T细胞和巨噬细胞募集到肾脏,促进免疫介导的肾脏疾病[90]。在基因敲除狼疮性肾炎小鼠模型(MRL/lpr)中,Th1和Th17细胞浸润到肾脏,使肾脏的形态和功能损伤受到抑制[91],表明CXCR3在狼疮性肾炎的形成过程中具有重要作用。Fan等[92]分析了健康女性和男性之间B细胞的差异表达基因,发现在不同性别间有358个差异表达基因;在女性中分别有226和132个基因上调和下调,通过qRT-PCR证实,位于前20个上调基因中。Im等[93]报道,内含子1中的rs34334103位点多态性与男性SLE患者相关。临床研究表明,在SLE患者中,表达显著增加;在患者肾活检中,约60%的浸润性T细胞表达,并且在患有狼疮性肾炎SLE患者的尿液中,可检测到阳性细胞;在肾脏受累的SLE患者中发现,细胞表面的表达水平升高[94]。Hewagama等[82]报道,用PCR阵列检测miR188-3p、和等X连锁基因,发现这些基因在女性SLE患者中的表达显著高于男性,同时,女性SLE患者的CD4+T细胞中,相同的mRNA和miRNA转录产物也被去甲基化和过表达,作者认为可能是由于女性SLE患者逃逸了失活,导致女性对SLE的易感性增加。
2.4 IRAK-1
(interleukin‑1 receptor‑associated kinase‑1,IRAK‑1)定位于Xq28,编码丝氨酸-苏氨酸激酶,参与TLR和IL-1受体信号通路,可激活许多下游衔接蛋白和蛋白激酶,从而激活NF-κB转录因子,介导免疫反应[95]。巨噬细胞中,NF-κB的激活,可促进TNF-α、IL-1β、IL-6、IL-12和趋化因子等促炎症细胞因子,诱导和放大炎症;T细胞中的NF-κB激活,可通过影响原始T细胞向Th1、Th2和Th17的分化来调节免疫反应,总之,IRAK-1通过促进各种T细胞亚群的分化和激活,促进自身免疫[96~98]。
在全基因组关联研究中,发现参与IFNα产生和/或反应性基因与SLE相关,包括和等。其中,特别令人感兴趣。首先,位于X染色体上,这使得其在研究性别依赖性疾病模式时成为一个引人注目的候选基因;其次,IRAK1蛋白是大多数TLR介导的细胞内信号传导所必需的,能够促进IFNα的分泌;最后,已被证明是SLE血清中IgG免疫复合物诱导的浆细胞样树突状细胞活化和IFNα产生所必需的[99]。2009年,Jacob等[100]分析了基因多态性与SLE的关联性,样本包括了769名儿童期发病的SLE患者、5337名北美成人发病的SLE患者和5317名健康对照,结果表明,有5个SNP位点与SLE相关联;随后,通过使用携带疾病基因座Sle1或Sle3的先天性小鼠模型,检测了的功能,发现的缺失可以导致IgM和IgG等自身抗体减少、抑制淋巴细胞活化,最终使所有狼疮相关的表型消失,证实是SLE的遗传易感因素,为SLE性别差异可能部分归因于X染色体连锁基因提供了依据。Li等[101]研究也证实,易患狼疮的B6.lpr小鼠脾脏单核细胞和SLE患者PBMC中的表达显著上调,SLE患者CD8+和Tregs中,尤其是Tregs中的表达也上调;重要的是,SLE患者中,Tregs中的表达水平与SLEDAI评分呈正相关,但与血清C3水平呈负相关,的表达水平与SLE患者Tregs凋亡的百分比呈正相关,表明,IRAK1是SLE患者自发Treg细胞凋亡的关键介质,特别是在肾病患者中[102]。
临床研究表明,在不同性别的SLE患者间的表达也有明显的差异,与男性SLE患者相比,女性SLE患者T细胞中的表达升高,而女性健康对照组与男性SLE患者的表达谱相似[64];SLE患者CD4+T细胞与健康对照相比,的表达显著增高,抑制SLE患者原始T细胞中的表达可阻止Th17分化,表明IRAK-1可能通过增强Th17应答而参与SLE的发病[103]。动物实验也得到了类似的结果,将雌性造血细胞移植到SLE杂交小鼠模型中,IFN-α的血清水平升高,而基因参与了IFN-α下游的信号通路,这些结果表明,在SLE性别二态性的形成过程中可能具有重要作用[104]。
2.5 CXorf21
(chromosome X open reading frame 21,)定位于Xp21,属于1型IFN诱导基因,启动子区有NF-κB、STAT1、STAT2、STAT3和IRF3转录因子的结合位点[85,105]。CXorf21作为TLR7、TLR8和TLR9下游的接头分子,可与 溶质载体蛋白SLC15A4 (solute carrier family 15,number 4,SLC15A4)结合形成复合物,特异性激活转录因子IRF5 (interferon regulator factor 5,IRF5)。敲除后,会增加雌性单核细胞、B细胞和DC的内溶酶体pH值,降低THP-1和pDC细胞系以及原代人单核细胞对TLR7和TLR9的反应,下调IFNα、TNFα、IL-6和各种趋化因子的表达,抑制STAT1和IRF5的磷酸化[105~108];此外,雌性APC的内溶酶体pH值低于雄性APC,这表明CXorf21可能调节抗原递呈,因为较低的内溶酶体pH值有利于内溶酶体室中的抗原处理,从而有利于随后抗原在细胞表面的呈递[107]。
2015年,Bentham等[109]在欧洲人中对7219例SLE病例和15,991名对照进行了大规模GWAS研究,确定了43个与SLE相关联的SNP位点,其中包括基因的rs887369位点。但Kwon等[110]在属于东亚人群的韩国人中并没有复制出这一结果。Zhang等[22]分析了从欧洲人和约鲁巴人的B淋巴细胞系获得的RNA和基因型测序数据,鉴定了114个逃逸基因,其中包括基因。Vawter等[111]通过Affymetrix U133P微阵列平台,对来自患有Klinefelter综合征(47,XXY)和对照男性(46,XY)淋巴母细胞样细胞系的mRNA进行了分析,发现Klinefelter综合征有129个差异表达基因,其中有14个X染色体基因(包括),这些基因在Klinefelter综合征患者中的表达显著升高,证实是一个X染色体失活逃逸基因。对SLE患者全血基因表达研究也证实,在女性SLE患者中显著上调,ROC曲线分析显示,AUC为92.6%,表明是与SLE密切相关的基因[112]。之后,Odhams等[108]报道,女性SLE患者免疫细胞中的水平显著高于男性SLE患者;流式细胞术观察到表达与疾病活动之间的年龄依赖性相关性,患者中CXorf21蛋白丰度与SLEDAI评分呈正相关;确定了基因启动子区有IRF3、NF-κB和STAT1-3的结合位点,表明转录可能是TLR4(IRF3)和IFN(STATs)信号通路的主要反应基因。证实在LPS和IFN-γ刺激的单核细胞以及IFN-α刺激的B细胞中的表达上调,女性的表达增加幅度更大,呈现出明显的性别二态性,这与先前已经报道的LPS诱导的单核细胞反应具有性别差异,即与雄性相比,雌性具有更高的激活和细胞因子释放的结果相一致。这些研究结果表明,是一个由IFN诱导的X染色体失活逃逸基因,为SLE性别二态性形成的遗传基础提供了证据。
3 结语与展望
大多数人类疾病具有性别间差异,其形成的机制十分复杂,涉及到性激素、肠道微生物以及X染色体失活等多种因素。近年来,失活X染色体上基因逃逸已成为人类疾病性别二态性研究的热点。由于哺乳类动物性染色体模式的形成,使X染色体携带的基因远多于Y染色体,因而,在进化过程中,许多物种形成了剂量补偿机制,通过X染色体失活平衡两性之间性染色体连锁基因的表达及其功能。越来越多的证据表明,Xi上存在许多能够逃逸失活的基因,这些基因不仅有物种间的差异,也存在着个体及组织间的异质性。尽管失活X染色体上基因逃逸机制的研究已取得了许多令人鼓舞的成果,发现了与基因逃逸相关的DNA、RNA、组蛋白的表观修饰、众多的调控蛋白以及染色质的空间结构的改变,然而,要完全解码失活X染色体基因逃逸的分子机制,还有很长的路。人类X染色体携带高密度的免疫相关基因,失活X染色体上免疫相关基因逃逸,一方面使女性获得生存优势,如妊娠成功、抗感染能力增强,但另一方面,也使女性对大多数自身免疫性疾病的易感性增加,或许这是自身免疫性疾病好发于女性的主要原因之一。SLE等自身免疫性疾病可作为研究失活X染色体上基因逃逸的病理模型。目前的研究提供了许多逃逸基因在自身免疫中的作用的证据,未来还需要更多的研究来评估这些基因在SLE患者以及动物模型中对女性偏好的贡献,筛选有价值的靶点以及针对这些分子治疗干预的疗效;同时,对失活X染色体上基因逃逸在自身免疫性疾病发病过程中作用的研究也将有助于进一步揭示基因逃逸及其异质性的分子机制。
[1] Zucchi D, Silvagni E, Elefante E, Signorini V, Cardelli C, Trentin F, Schilirò D, Cascarano G, Valevich A, Bortoluzzi A, Tani C. Systemic lupus erythematosus: one year in review 2023., 2023, 41(5): 997–1008.
[2] Tian JR, Zhang DY, Yao X, Huang YQ, Lu QJ. Global epidemiology of systemic lupus erythematosus: a comprehensive systematic analysis and modelling study., 2023, 82(3): 351–356.
[3] Ortona E, Pierdominici M, Maselli A, Veroni C, Aloisi F, Shoenfeld Y. Sex-based differences in autoimmune diseases., 2016, 52(2): 205–212.
[4] Nusbaum JS, Mirza I, Shum J, Freilich RW, Cohen RE, Pillinger MH, Izmirly PM, Buyon JP. Sex differences in systemic lupus erythematosus: epidemiology, clinical considerations, and disease pathogenesis.,2020, 95(2): 384–394.
[5] Christou EAA, Banos A, Kosmara D, Bertsias GK, Boumpas DT. Sexual dimorphism in SLE: above and beyond sex hormones., 2019, 28(1): 3–10.
[6] Posynick BJ, Brown CJ. Escape from X-chromosome inactivation: an evolutionary perspective.,2019, 7: 241.
[7] Loda A, Collombet S, Heard E. Gene regulation in time and space during X-chromosome inactivation.,2022, 23(4): 231–249.
[8] Berletch JB, Yang F, Disteche CM. Escape from X inactivation in mice and humans.,2010, 11(6): 213.
[9] Balaton BP, Brown CJ. Escape artists of the X chromosome., 2016, 32(6): 348–359.
[10] Lyon M F. Genetic activity of sex chromosomes in somatic cells of mammals., 1970, 259(828): 41–52.
[11] Schempp W, Meer B. Cytologic evidence for three human X-chromosomal segments escaping inactivation.,1983, 63(2): 171–174.
[12] Disteche CM. Escapees on the X chromosome., 1999, 96(25): 14180–14182.
[13] Carrel L, Willard HF. Heterogeneous gene expression from the inactive X chromosome: an X-linked gene that escapes X inactivation in some human cell lines but is inactivated in others., 1999, 96(13): 7364–7369.
[14] Carrel L, Willard HF. X-inactivation profile reveals extensive variability in X-linked gene expression in females., 2005, 434(7031): 400–404.
[15] Carrel L, Brown CJ. When the Lyonized (ized chromosome) roars: ongoing expression from an inactive X chromosome.,2017, 372(1733): 20160355.
[16] Qi SH, Al Mamun A, Ngwa C, Romana S, Ritzel R, Arnold AP, McCullough LD, Liu FD. X chromosome escapee genes are involved in ischemic sexual dimorphism through epigenetic modification of inflammatory signals., 2021, 18(1): 70.
[17] Al Nadaf S, Deakin JE, Gilbert C, Robinson TJ, Graves JAM, Waters PD. A cross-species comparison of escape from X inactivation in Eutheria: implications for evolution of X chromosome inactivation., 2012, 121(1): 71–78.
[18] Yang F, Babak T, Shendure J, Disteche CM. Global survey of escape from X inactivation by RNA- sequencing in mouse., 2010, 20(5): 614–622.
[19] Wang X, Douglas KC, Vandeberg JL, Clark AG, Samollow PB. Chromosome-wide profiling of X-chromosome inactivation and epigenetic states in fetal brain and placenta of the opossum, Monodelphis domestica., 2014, 24(1): 70–83.
[20] Whitworth D J, Pask AJ. The X factor: X chromosome dosage compensation in the evolutionarily divergent monotremes and marsupials., 2016, 56: 117–121.
[21] Balaton BP, Cotton AM, Brown CJ. Derivation of consensus inactivation status for X-linked genes from genome-wide studies., 2015, 6: 35.
[22] Zhang YC, Castillo-Morales A, Jiang M, Zhu YF, Hu LD, Urrutia AO, Kong XY, Hurst LD. Genes that escape X-inactivation in humans have high intraspecific variability in expression,are associated with mental impairment but are not slow evolving.,2013, 30(12): 2588–2601.
[23] Tukiainen T, Villani AC, Yen A, Rivas MA, Marshall JL, Satija R, Aguirre M, Gauthier L, Fleharty M, Kirby A, Cummings BB, Castel SE, Karczewski KJ, Aguet F, Byrnes A, GTEx Consortium, Laboratory, Data Analysis & Coordinating Center (LDACC)—Analysis Working Group, Statistical Methods groups—Analysis Working Group, Enhancing GTEx (eGTEx) groups, NIH Common Fund, NIH/NCI, NIH/NHGRI, NIH/NIMH , NIH/NIDA, Biospecimen Collection Source Site—NDRI, Biospecimen Collection Source Site—RPCI, Biospecimen Core Resource—VARI, Brain Bank Repository—University of Miami Brain Endowment Bank, Leidos Biomedical— Project Management, ELSI Study, Genome Browser Data Integration &Visualization—EBI, Genome Browser Data Integration &Visualization—UCSC Genomics Institute, University of California Santa Cruz, Lappalainen T, Regev A, Ardlie KG, Hacohen N, MacArthur DG. Landscape of X chromosome inactivation across human tissues., 2017, 550: 244–248.
[24] Garieri M, Stamoulis G, Blanc X, Falconnet E, Ribaux P, Borel C, Santoni F, Antonarakis SE. Extensive cellular heterogeneity of X inactivation revealed by single-cell allele-specific expression in human fibroblasts., 2018, 115(51): 13015–13020.
[25] Zito A, Roberts AL, Visconti A, Rossi N, Andres-Ejarque R, Nardone S, El-Sayed Moustafa JS, Falchi M, Small KS. Escape from X-inactivation in twins exhibits intra- and inter-individual variability across tissues and is heritable., 2023, 19(2): e1010556.
[26] Libert C, Dejager L, Pinheiro I. The X chromosome in immune functions: when a chromosome makes the difference., 2010, 10(8): 594–604.
[27] Neri G, Schwartz CE, Lubs HA, Stevenson RE. X-linked intellectual disability update 2017., 2018, 176(6): 1375–1388.
[28] Dunford A, Weinstock DM, Savova V, Schumacher SE, Cleary JP, Yoda A, Sullivan TJ, Hess JM, Gimelbrant AA, Beroukhim R, Lawrence MS, Getz G, Lane AA. Tumor-suppressor genes that escape from X-inactivation contribute to cancer sex bias., 2017, 49(1): 10–16.
[29] Clement-Jones M, Schiller S, Rao E, Blaschke RJ, Zuniga A, Zeller R, Robson SC, Binder G, Glass I, Strachan T, Lindsay S, Rappold GA. The short stature homeobox gene SHOX is involved in skeletal abnormalities in Turner syndrome., 2000, 9(5): 695–702.
[30] Sauteraud R, Stahl JM, James J, Englebright M, Chen F, Zhan XW, Carrel L, Liu DJ. Inferring genes that escape X-Chromosome inactivation reveals important contribution of variable escape genes to sex-biased diseases., 2021, 31(9): 1629–1637.
[31] Augui S, Nora EP, Heard E. Regulation of X-chromosome inactivation by the X-inactivation centre., 2011, 12(6): 429–442.
[32] Pandya-Jones A, Markaki Y, Serizay J, Chitiashvili T, Mancia Leon WR, Damianov A, Chronis C, Papp B, Chen CK, McKee R, Wang XJ, Chau A, Sabri S, Leonhardt H, Zheng SK, Guttman M, Black DL, Plath K. A protein assembly mediates Xist localization and gene silencing., 2020, 587(7832): 145–151.
[33] Zhou VW, Goren A, Bernstein BE. Charting histone modifications and the functional organization of mammalian genomes., 2011, 12(1): 7–18.
[34] Loda A, Heard E. Xist RNA in action: past, present, and future., 2019, 15: e1008333.
[35] Wang ZZ, Cai HR, Zhao EH, Cui HJ. The diverse roles of histone demethylase KDM4B in normal and cancer development and progression., 2022, 9: 790129.
[36] Sadreyev RI, Yildirim E, Pinter SF, Lee JT. Bimodal quantitative relationships between histone modifications for X-linked and autosomal loci.,2013, 110(17): 6949–6954.
[37] Kelsey AD, Yang C, Leung D, Minks J, Dixon- McDougall T, Baldry SEL, Bogutz AB, Lefebvre L, Brown CJ. Impact of flanking chromosomal sequences on localization and silencing by the human non-coding RNA XIST., 2015, 16: 208.
[38] Brockdorff N. Polycomb complexes in X chromosome inactivation., 2017, 372(1733): 20170021.
[39] Cerase A, Smeets D, Tang YA, Gdula M, Kraus F, Spivakov M, Moindrot B, Leleu M, Tattermusch A, Demmerle J, Nesterova TB, Green C, Otte AP, Schermelleh L, Brockdorff N. Spatial separation of Xist RNA and polycomb proteins revealed by superresolution microscopy., 2014, 111(6): 2235–2240.
[40] Simon MD, Pinter SF, Fang R, Sarma K, Rutenberg- Schoenberg M, Bowman SK, Kesner BA, Maier VK, Kingston RE, Lee JT. High-resolution Xist binding maps reveal two-step spreading during X-chromosome inactivation., 2013, 504(7480): 465–469.
[41] Brinkman AB, Roelofsen T, Pennings SW, Martens JHA, Jenuwein T, Stunnenberg HG. Histone modification patterns associated with the human X chromosome., 2006, 7(6): 628–634.
[42] Tang SJ. Chromatin organization by repetitive elements (CORE): a genomic principle for the higher-order structure of chromosomes., 2011, 2: 502–515.
[43] Wang Z, Willard HF, Mukherjee S, Furey TS. Evidence of influence of genomic DNA sequence on human X chromosome inactivation., 2006, 2(9): e113.
[44] Lyon MF. X-chromosome inactivation: a repeat hypothesis., 1998, 80(1–4): 133–137.
[45] Ross MT, Grafham DV, Coffey AJ, Scherer S, McLay K, Muzny D, Platzer M, Howell GR, Burrows C, Bird CP, Frankish A, Lovell FL, Howe KL, Ashurst JL, Fulton RS, Sudbrak R, Wen GP, Jones MC, Hurles ME, Andrews TD, Scott CE, Searle S, Ramser J, Whittaker A, Deadman R, Carter NP, Hunt SE, Chen R, Cree A, Gunaratne P, Havlak P, Hodgson A, Metzker ML, Richards S, Scott G, Steffen D, Sodergren E, Wheeler DA, Worley KC, Ainscough R, Ambrose KD, Ansari-Lari MA, Aradhya S, Ashwell RIS, Babbage AK, Bagguley CL, Ballabio A, Banerjee R, Barker GE, Barlow KF, Barrett IP, Bates KN, Beare DM, Beasley H, Beasley O, Beck A, Bethel G, Blechschmidt K, Brady N, Bray-Allen S, Bridgeman AM, Brown AJ, Brown MJ, Bonnin D, Bruford EA, Buhay C, Burch P, Burford D, Burgess J, Burrill W, Burton J, Bye JM, Carder C, Carrel L, Chako J, Chapman JC, Chavez D, Chen E, Chen G, Chen Y, Chen ZJ, Chinault C, Ciccodicola A, Clark SY, Clarke G, Clee CM, Clegg S, Clerc-Blankenburg K, Clifford K, Cobley V, Cole CG, Conquer JS, Corby N, Connor RE, David R, Davies J, Davis C, Davis J, Delgado O, Deshazo D, Dhami P, Ding Y, Dinh H, Dodsworth S, Draper H, Dugan-Rocha S, Dunham A, Dunn M, Durbin KJ, Dutta I, Eades T, Ellwood M, Emery-Cohen A, Errington H, Evans KL, Faulkner L, Francis F, Frankland J, Fraser AE, Galgoczy P, Gilbert J, Gill R, Glöckner G, Gregory SG, Gribble S, Griffiths C, Grocock R, Gu Y, Gwilliam R, Hamilton C, Hart EA, Hawes A, Heath PD, Heitmann K, Hennig S, Hernandez J, Hinzmann B, Ho S, Hoffs M, Howden PJ, Huckle EJ, Hume J, Hunt PJ, Hunt AR, Isherwood J, Jacob L, Johnson D, Jones S, de Jong PJ, Joseph SS, Keenan S, Kelly S, Kershaw JK, Khan Z, Kioschis P, Klages S, Knights AJ, Kosiura A, Kovar-Smith C, Laird GK, Langford C, Lawlor S, Leversha M, Lewis L, Liu W, Lloyd C, Lloyd DM, Loulseged H, Loveland JE, Lovell JD, Lozado R, Lu J, Lyne R, Ma J, Maheshwari M, Matthews LH, McDowall J, McLaren S, McMurray A, Meidl P, Meitinger T, Milne S, Miner G, Mistry SL, Morgan M, Morris S, Müller I, Mullikin JC, Nguyen N, Nordsiek G, Nyakatura G, O'Dell CN, Okwuonu G, Palmer S, Pandian R, Parker D, Parrish J, Pasternak S, Patel D, Pearce AV, Pearson DM, Pelan SE, Perez L, Porter KM, Ramsey Y, Reichwald K, Rhodes S, Ridler KA, Schlessinger D, Schueler MG, Sehra HK, Shaw-Smith C, Shen H, Sheridan EM, Shownkeen R, Skuce CD, Smith ML, Sotheran EC, Steingruber HE, Steward CA, Storey R, Swann RM, Swarbreck D, Tabor PE, Taudien S, Taylor T, Teague B, Thomas K, Thorpe A, Timms K, Tracey A, Trevanion S, Tromans AC, d'Urso M, Verduzco D, Villasana D, Waldron L, Wall M, Wang Q, Warren J, Warry GL, Wei X, West A, Whitehead SL, Whiteley MN, Wilkinson JE, Willey DL, Williams G, Williams L, Williamson A, Williamson H, Wilming L, Woodmansey RL, Wray PW, Yen J, Zhang J, Zhou J, Zoghbi H, Zorilla S, Buck D, Reinhardt R, Poustka A, Rosenthal A, Lehrach H, Meindl A, Minx PJ, Hillier LW, Willard HF, Wilson RK, Waterston RH, Rice CM, Vaudin M, Coulson A, Nelson DL, Weinstock G, Sulston JE, Durbin R, Hubbard T, Gibbs RA, Beck S, Rogers J, Bentley DR. The DNA sequence of the human X chromosome., 2005, 434: 325–337.
[46] Chow JC, Ciaudo C, Fazzari MJ, Mise N, Servant N, Glass JL, Attreed M, Avner P, Wutz A, Barillot E, Greally JM, Voinnet O, Heard E. LINE-1 activity in facultative heterochromatin formation during X chromosome inactivation., 2016, 166(3): 782.
[47] Li SY, Shen XH. Long interspersed nuclear element 1 and B1/Alu repeats blueprint genome compartmentalization.,2023, 80: 102049.
[48] Nguyen DK, Yang F, Kaul R, Alkan C, Antonellis A, Friery KF, Zhu BL, de Jong PJ, Disteche CM. Clcn4–2 genomic structure differs between the X locus in Mus spretus and the autosomal locus in: AT motif enrichment on the X., 2011, 21(3): 402–409.
[49] Helbig R, Fackelmayer FO. Scaffold attachment factor A (SAF-A) is concentrated in inactive X chromosome territories through its RGG domain., 2003, 112(4): 173–182.
[50] Agrelo R, Souabni A, Novatchkova M, Haslinger C, Leeb M, Komnenovic V, Kishimoto H, Gresh L, Kohwi- Shigematsu T, Kenner L, Wutz A. SATB1 defines the developmental context for gene silencing by Xist in lymphoma and embryonic cells., 2009, 16(4): 507–516.
[51] Kohwi-Shigematsu T, Kohwi Y, Takahashi K, Richards HW, Ayers SD, Han HJ, Cai ST. SATB1-mediated functional packaging of chromatin into loops., 2012, 58(3): 243–254.
[52] Galande S, Purbey PK, Notani D, Kumar PP. The third dimension of gene regulation: organization of dynamic chromatin loopscape by SATB1., 2007, 17(5): 408–414.
[53] Hasegawa Y, Brockdorff N, Kawano S, Tsutui K, Tsutui K, Nakagawa S. The matrix protein hnRNP U is required for chromosomal localization of Xist RNA., 2010, 19(3): 469–476.
[54] Sanborn AL, Rao SS, Huang SC, Durand NC, Huntley MH, Jewett AI, Bochkov ID, Chinnappan D, Cutkosky A, Li J, Geeting KP, Gnirke A, Melnikov A, McKenna D, Stamenova EK, Lander ES, Aiden EL. Chromatin extrusion explains key features of loop and domain formation in wild-type and engineered genomes., 2015, 112(47): E6456–E6465.
[55] Filippova GN, Cheng MK, Moore JM, Truong JP, Hu YJ, Nguyen DK, Tsuchiya KD, Disteche CM. Boundaries between chromosomal domains of X inactivation and escape bind CTCF and lack CpG methylation during early development., 2005, 8(1): 31–42.
[56] Fang H, Tronco AR, Bonora G, Nguyen T, Thakur J, Berletch JB, Filippova GN, Henikoff S, Shendure J, Noble WS, Disteche CM, Deng XX. CTCF-mediated insulation and chromatin environment modulate Car5b escape from X inactivation., 2023.
[57] Chen CY, Shi WQ, Balaton BP, Matthews AM, Li YF, Arenillas DJ, Mathelier A, Itoh M, Kawaji H, Lassmann T, Hayashizaki Y, Carninci P, Forrest ARR, Brown CJ, Wasserman WW. YY1 binding association with sex- biased transcription revealed through X-linked transcript levels and allelic binding analyses., 2016, 6: 37324.
[58] Li L, Williams P, Ren WD, Wang MY, Gao Z, Miao WL, Huang M, Song JK, Wang YS. YY1 interacts with guanine quadruplexes to regulate DNA looping and gene expression., 2021, 17(2): 161–168.
[59] Sun S, Del Rosario BC, Szanto A, Ogawa Y, Jeon Y, Lee JT. Jpx RNA activates Xist by evicting CTCF., 2013, 153(7): 1537–1551.
[60] Oh HJ, Aguilar R, Kesner B, Lee HG, Kriz AJ, Chu HP, Lee JT. Jpx RNA regulates CTCF anchor site selection and formation of chromosome loops., 2021, 184(25): 6157–6173.e24.
[61] Jiwrajka N, Anguera MC. The X in sex-biased immunity and autoimmune rheumatic disease., 2022, 219(6): e20211487.
[62] Miquel CH, Faz-Lopez B, Guéry JC. Influence of X chromosome in sex-biased autoimmune diseases., 2023, 137: 102992.
[63] Berletch JB, Yang F, Xu J, Carrel L, Disteche CM. Genes that escape from X inactivation., 2011, 130(2): 237–245.
[64] Sarmiento L, Svensson J, Barchetta I, Mousavi MJ, Mahmoudi M, Ghotloo S. Escape from X chromosome inactivation and female bias of autoimmune diseases., 2020, 26(1): 127.
[65] Syrett CM, Anguera MC. When the balance is broken: X linked gene dosage from two X chromosomes and female-biased autoimmunity., 2019, 106(4): 919–932.
[66] Sarmiento L, Svensson J, Barchetta I, Giwercman A, Cilio CM. Copy number of the X-linked genes TLR7 and CD40L influences innate and adaptive immune responses., 2019, 90(2): e12776.
[67] Souyris M, Cenac C, Azar P, Daviaud D, Canivet A, Grunenwald S, Pienkowski C, Chaumeil J, Mejía J, Guéry JC. TLR7 escapes X chromosome inactivation in immune cells., 2018, 3(19): eaap8855.
[68] Fillatreau S, Manfroi B, Dörner T. Toll-like receptor signalling in B cells during systemic lupus erythematosus., 2021, 17(2): 98–108.
[69] Souyris M, Mejía JE, Chaumeil J, Guéry JC. Female predisposition to TLR7-driven autoimmunity: gene dosage and the escape from X chromosome inactivation.,2019, 41(2): 153–164.
[70] Murphy ED, Roths JB. A Y chromosome associated factor in strain BXSB producing accelerated autoimmunity and lymphoproliferation., 1979, 22(11): 1188–1194.
[71] Merino R, Fossati L, Izui S. The lupus-prone BXSB strain: the Yaa gene model of systemic lupus erythematosus., 1992, 14(2): 141–157.
[72] Izui S, Iwamoto M, Fossati L, Merino R, Takahashi S, Ibnou-Zekri N. The Yaa gene model of systemic lupus erythematosus., 1995, 144: 137–156.
[73] Amano H, Amano E, Moll T, Marinkovic D, Ibnou-Zekri N, Martinez-Soría E, Semac I, Wirth T, Nitschke L, Izui S. The Yaa mutation promoting murine lupus causes defective development of marginal zone B cells., 2003, 170(5): 2293–2301.
[74] Pisitkun P, Deane JA, Difilippantonio MJ, Tarasenko T, Satterthwaite AB, Bolland S. Autoreactive B cell responses to RNA-related antigens due to TLR7 gene duplication., 2006, 312(5780): 1669–1672.
[75] Slae M, Heshin-Bekenstein M, Simckes A, Heimer G, Engelhard D, Eisenstein EM. Female polysomy-X and systemic lupus erythematosus., 2014, 43(4): 508–512.
[76] Harris VM, Sharma R, Cavett J, Kurien BT, Liu K, Koelsch KA, Rasmussen A, Radfar L, Lewis D, Stone DU, Kaufman CE, Li SB, Segal B, Wallace DJ, Weisman MH, Venuturupalli S, Kelly JA, Alarcon-Riquelme ME, Pons-Estel B, Jonsson R, Lu XL, Gottenberg JE, Anaya JM, Cunninghame-Graham DS, Huang AJW, Brennan MT, Hughes P, Alevizos I, Miceli-Richard C, Keystone EC, Bykerk VP, Hirschfield G, Xie G, Siminovitch KA, Ng WF, Nordmark G, Bucher SM, Eriksson P, Omdal R, Rhodus NL, Rischmueller M, Rohrer M, Wahren- Herlenius M, Witte T, Mariette X, Lessard CJ, Harley JB, Sivils KL, Scofield RH. Klinefelter’s syndrome (47,XXY) is in excess among men with Sjögren’s syndrome., 2016, 168: 25–29.
[77] Sawalha AH, Harley JB, Scofield RH. Autoimmunity and Klinefelter’s syndrome: when men have two X chromosomes., 2009, 33(1): 31–34.
[78] Invernizzi P, Miozzo M, Oertelt-Prigione S, Meroni PL, Persani L, Selmi C, Battezzati PM, Zuin M, Lucchi S, Marasini B, Zeni S, Watnik M, Tabano S, Maitz S, Pasini S, Gershwin ME, Podda M. X monosomy in female systemic lupus erythematosus., 2007, 1110: 84–91.
[79] Liu K, Kurien BT, Zimmerman SL, Kaufman KM, Taft DH, Kottyan LC, Lazaro S, Weaver CA, Ice JA, Adler AJ, Chodosh J, Radfar L, Rasmussen A, Stone DU, Lewis DM, Li SB, Koelsch KA, Igoe A, Talsania M, Kumar J, Maier-Moore JS, Harris VM, Gopalakrishnan R, Jonsson R, Lessard JA, Lu XL, Gottenberg JE, Anaya JM, Cunninghame-Graham DS, Huang AJW, Brennan MT, Hughes P, Illei GG, Miceli-Richard C, Keystone EC, Bykerk VP, Hirschfield G, Xie G, Ng WF, Nordmark G, Eriksson P, Omdal R, Rhodus NL, Rischmueller M, Rohrer M, Segal BM, Vyse TJ, Wahren-Herlenius M, Witte T, Pons-Estel B, Alarcon-Riquelme ME, Guthridge JM, James JA, Lessard CJ, Kelly JA, Thompson SD, Gaffney PM, Montgomery CG, Edberg JC, Kimberly RP, Alarcón GS, Langefeld CL, Gilkeson GS, Kamen DL, Tsao BP, McCune WJ, Salmon JE, Merrill JT, Weisman MH, Wallace DJ, Utset TO, Bottinger EP, Amos CI, Siminovitch KA, Mariette X, Sivils KL, Harley JB, Scofield RH. X Chromosome dose and sex bias in autoimmune diseases: increased prevalence of 47,XXX in systemic lupus erythematosus and Sjögren’s syndrome., 2016, 68(5): 1290–1300.
[80] Koshy M, Berger D, Crow MK. Increased expression of CD40 ligand on systemic lupus erythematosus lymphocytes., 1996, 98: 826–837.
[81] Desai-Mehta A, Lu L, Ramsey-Goldman R, Datta SK. Hyperexpression of CD40 ligand by B and T cells in human lupus and its role in pathogenic autoantibody production., 1996, 97(7): 2063–2073.
[82] Hewagama A, Gorelik G, Patel D, Liyanarachchi P, Liyanarachchi P, McCune WJ, Somers E, Gonzalez- Rivera T, Michigan Lupus Cohort, Strickland F, Richardson B. Overexpression of X-linked genes in T cells from women with lupus.,2013, 41: 60–71.
[83] Ramanujam M, Steffgen J, Visvanathan S, Mohan C, Fine JS, Putterman C. Phoenix from the flames: rediscovering the role of the CD40-CD40L pathway in systemic lupus erythematosus and lupus nephritis., 2020, 19(11): 102668.
[84] Vordenbäumen S, Sokolowski A, Rosenbaum A, Gebhard C, Raithel J, Düsing C, Chehab G, Richter JG, Brinks R, Rehli M, Schneider M. Methyl donor micronutrients, CD40-ligand methylation and disease activity in systemic lupus erythematosus: a cross-sectional association study., 2021, 30(11): 1773–1780.
[85] Lu QJ, Wu AL, Tesmer L, Ray D, Yousif N, Richardson B. Demethylation of CD40LG on the inactive X in T cells from women with lupus., 2007, 179(9): 6352–6358.
[86] Lacotte S, Brun S, Muller S, Dumortier H. CXCR3, inflammation, and autoimmune diseases., 2009, 1173: 310–317.
[87] Oghumu S, Varikuti S, Stock JC, Volpedo G, Saljoughian N, Terrazas CA, Satoskar AR. Cutting Edge: CXCR3 escapes X chromosome inactivation in T cells during infection: potential implications for sex differences in immune responses., 2019, 203(4): 789–794.
[88] Barbi J, Oghumu S, Lezama-Davila CM, Satoskar AR. IFN-gamma and STAT1 are required for efficient induction of CXC chemokine receptor 3 (CXCR3) on CD4+but not CD8+T cells., 2007, 110: 2215–2216.
[89] Wang GJ, Sun Y, Jiang YS, Li SZ, Liu YH, Yuan YY, Nie H. CXCR3 deficiency decreases autoantibody production by inhibiting aberrant activated T follicular helper cells and B cells in lupus mice., 2023, 156: 39–47.
[90] Menke J, Zeller GC, Kikawada E, Means TK, Huang XR, Lan HY, Lu B, Farber J, Luster AD, Kelley VR. CXCL9, but not CXCL10, promotes CXCR3-dependent immune- mediated kidney disease., 2008, 19: 1177–1189.
[91] Steinmetz OM, Turner JE, Paust HJ, Lindner M, Peters A, Heiss K, Velden J, Hopfer H, Fehr S, Krieger T, Mey er-Schwesinger C, Meyer TN, Helmchen U, Mittrücker HW, Stahl RAK, Panzer U. CXCR3 mediates renal Th1 and Th17 immune response in murine lupus nephritis., 2009, 183: 4693–4704.
[92] Fan HY, Dong GJ, Zhao GF, Liu F, Yao GH, Zhu YC, Hou YJ. Gender differences of B cell signature in healthy subjects underlie disparities in incidence and course of SLE related to estrogen., 2014, 2014: 814598.
[93] Im CH, Park JA, Kim JY, Lee EY, Lee EB, Kim Y, Song YW. CXCR3 polymorphism is associated with male gender and pleuritis in patients with systemic lupus erythematosus., 2014, 75(5): 466-469.
[94] Enghard P, Humrich JY, Rudolph B, Rosenberger S, Biesen R, Kuhn A, Manz R, Hiepe F, Radbruch A, Burmester GR, Riemekasten G. CXCR3+CD4+T cells are enriched in inflamed kidneys and urine and provide a new biomarker for acute nephritis flares in systemic lupus erythematosus patients., 2009, 60: 199–206.
[95] Cooke EL, Uings IJ, Xia CL, Woo P, Ray KP. Functional analysis of the interleukin-1-receptor-associated kinase (IRAK-1) in interleukin-1 beta-stimulated nuclear factor kappa B (NF-kappa B) pathway activation: IRAK-1 associates with the NF-kappa B essential modulator (NEMO) upon receptor stimulation., 2001, 359: 403–410.
[96] Li-Weber M, Giaisi M, Baumann S, Pálf K, Krammer PH. NF-kappa B synergizes with NF-AT and NF-IL6 in activation of the IL-4 gene in T cells.,2004, 34: 1111–1118.
[97] Ruan QG, Kameswaran V, Zhang Y, Zheng SJ, Sun J, Wang JM, DeVirgiliis J, Liou HC, Beg AA, Chen YH. The Th17 immune response is controlled by the Rel-RORγ-RORγ T transcriptional aXis., 2011, 208: 2321–2333.
[98] Oh H, Ghosh S. NF-kappa B: roles and regulation in different CD4(+) T-cell subsets., 2013, 252: 41–51.
[99] Bronson PG, Chaivorapol C, Ortmann W, Behrens TW, Graham RR. The genetics of type I interferon in systemic lupus erythematosus., 2012, 24(5): 530–537.
[100] Jacob CO, Zhu JK, Armstrong DL, Yan M, Han J, Zhou XJ, Thomas JA, Reiff A, Myones BL, Ojwang JO, Kaufman KM, Klein-Gitelman M, McCurdy D, Wagner-Weiner L, Silverman E, Ziegler J, Kelly JA, Merrill JT, Harley JB, Ramsey-Goldman R, Vila LM, Bae SC, Vyse TJ, Gilkeson GS, Gaffney PM, Moser KL, Langefeld CD, Zidovetzki R, Mohan C. Identifciation of IRAK1 as a risk gene with critical role in the pathogenesis of systemic lupus erythematosus.,2009, 106: 6256–6261.
[101] Li MF, Yu DT, Ni B, Hao F. Interleukin-1 receptor associated kinase 1 is a potential therapeutic target of anti-inflammatory therapy for systemic lupus erythematosus., 2017, 87: 94–101.
[102] Li MF, Yu DT, Wang Y, Luo N, Han GM, Yang B. Interferon-α activates interleukin-1 receptor-associated kinase 1 to induce regulatory T-cell apoptosis in patients with systemic lupus erythematosus., 2021, 48(8): 1172–1185.
[103] Zhou Z, Tian ZQ, Zhang MJ, Zhang YX, Ni B, Hao F. Upregulated IL-1 receptor-associated kinase 1 (IRAK1) in systemic lupus erythematosus: IRAK1 inhibition represses Th17 differentiation with therapeutic potential.,2018, 47(5): 468–483.
[104] David A, Trigunaite A, Macleod MK, Johnson AC, Marrack P, Jørgensen TN. Intrinsic autoimmune capacities of hematopoietic cells from female New Zealand hybrid mice., 2014, 15(3): 153–161.
[105] Odhams CA, Roberts AL, Vester SK, Duarte CST, Beales CT, Clarke AJ, Lindinger S, Daffern SJ, Zito A, Chen LY, Jones LL, Boteva L, Morris DL, Small KS, Fernando MMA, Cunninghame Graham DS, Vyse TJ. Interferon inducible X-linked gene CXorf21 may contribute to sexual dimorphism in Systemic Lupus Erythematosus., 2019, 10(1): 2164.
[106] Harris VM, Harley ITW, Kurien BT, Koelsch KA, Scofield RH. Lysosomal pH is regulated in a sex dependent manner in immune cells expressing CXorf21., 2019, 10: 578.
[107] Harris VM, Koelsch KA, Kurien BT, Harley ITW, Wren JD, Harley JB, Scofield RH. Characterization of cxorf21 provides molecular insight into female-bias immune response in SLE pathogenesis., 2019, 10: 2160.
[108] Heinz LX, Lee J, Kapoor U, Kartnig F, Sedlyarov V, Papakostas K, César-Razquin A, Essletzbichler P, Goldmann U, Stefanovic A, Bigenzahn JW, Scorzoni S, Pizzagalli MD, Bensimon A, Müller AC, King FJ, Li J, Girardi E, Mbow ML, Whitehurst CE, Rebsamen M, Superti-Furga G. TASL is the SLC15A4-associated adaptor for IRF5 activation by TLR7-9., 2020, 581(7808): 316–322.
[109] Bentham J, Morris DL, Graham DSC, Pinder CL, Tombleson P, Behrens TW, Martín J, Fairfax BP, Knight JC, Chen LY, Replogle J, Syvänen AC, Rönnblom L, Graham RR, Wither JE, Rioux JD, Alarcón-Riquelme ME, Vyse TJ. Genetic association analyses implicate aberrant regulation of innate and adaptive immunity genes in the pathogenesis of systemic lupus erythematosus., 2015, 47(12): 1457–1464.
[110] Kwon KS, Cho HY, Chung YJ. Recapitulation of candidate systemic lupus erythematosus-associated variants in Koreans., 2016, 14: 85–89.
[111] Vawter MP, Harvey PD, DeLisi LE. Dysregulation of X-linked gene expression in Klinefelter’s syndrome and association with verbal cognition., 2007, 144: 728–734 .
[112] Mackay M, Oswald M, Sanchez-Guerrero J, Lichauco J, Aranow C, Kotkin S, Korsunsky I, Gregersen PK, Diamond B. Molecular signatures in systemic lupus erythematosus: distinction between disease flare and infection., 2016, 3: e000159.
Genes that escape from X-chromosome inactivation and sexual dimorphism of systemic lupus erythematosus
Qian Ma1,2, Shaolan Zhou3, Jie Dang2,4, Zhenghao Huo2,4, Zhanbing Ma2,4
X chromosome inactivation can balance the effects of the two X chromosomes in females, and emerging evidence indicates that numerous genes on the inactivated X chromosome have the potential to evade inactivation. The mechanisms of escape include modification of DNA, RNA, histone, epitope, and various regulatory proteins, as well as the spatial structure of chromatin. The study of X chromosome inactivation escape has paved the way for investigating sex dimorphism in human diseases, particularly autoimmune diseases. It has been demonstrated that the presence of,,,, andsignificantly contributes to the prevalence of SLE (systemic lupus erythematosus) in females. This article mainly reviews the molecular mechanisms underlying these genes that escape from X-chromosome inactivation and sexual dimorphism of systemic lupus erythematosus. Therefore, elucidating the molecular mechanisms underlying sexual dimorphism in SLE is not only crucial for diagnosing and treating the disease, but also holds theoretical significance in comprehensively understanding the development and regulatory mechanisms of the human immune system.
inactive X chromosome; gene; escape; sexual dimorphism; systemic lupus erythematosus
2023-10-07;
2023-12-11;
2023-12-13
国家自然科学基金项目(编号:82060301, 81960306)和宁夏自然科学基金项目(编号:22KJB180014, 2020AAC03116)资助[Supported by the National Natural Science Foundation of China (Nos. 82060301, 81960306) and the Natural Science Foundation of the Ningxia (Nos. 22KJB180014, 2020AAC03116)]
马茜,博士,实验师,研究方向:病原生物学与宿主免疫。E-mail: maqian226@126.com
周少岚,博士,主治医师,研究方向:自身免疫性疾病的遗传学研究。E-mail: 64758961@qq.com
马茜和周少岚并列第一作者。
霍正浩,教授,研究方向:自身免疫性疾病的遗传学研究。E-mail: huozhh@163.com
马占兵,博士,副教授,研究方向:自身免疫性疾病的遗传学研究。E-mail: 1784947489@qq.com
10.16288/j.yczz.23-214
(责任编委: 谢小冬)
