Assessment of gastric cancer prognosis, immune infiltration based on cuproptosis-related LncRNAs and prediction of traditional Chinese medicine
2024-01-22YUNZhangjunSHENYangMISuicaiLIUZhuFANQiuyueLIUAiqiZHAIMiaojieHOULi
YUN Zhang-jun, SHEN Yang, MI Sui-cai, LIU Zhu, FAN Qiu-yue, LIU Ai-qi, ZHAI Miao-jie, HOU Li✉
1.Dongzhimen Hospital, Beijing University of Chinese Medicine, Beijing 100700, China
2.Department of Oncology, Xiamen Hospital of Traditional Chinese Medicine, Xiamen 361015, China
Keywords:
ABSTRACT
1.Introduction
Gastric cancer (GC) is malignant tumor that originates in the gastric epithelium.According to the latest cancer statistics from the World Health Organization, the number of new cases of GC in 2020 will be 1,089,000 and the number of deaths will be 769,000, with the 6th highest incidence rate and the 3rd highest mortality rate of global malignant tumors[1].China is a large country with GC, and about half of the new and fatal cases in the world will be in China in 2020, seriously threatening the life and health of the nation.The lack of specific symptoms in the early stage of GC increases the difficulty of diagnosis and treatment of GC.Clinical staging can assess the prognosis of patients to a certain extent and help them choose appropriate treatment strategies, but its accuracy still needs to be improved.Therefore, it is necessary to further explore biomarkers with higher prognostic efficacy, and to identify high-risk patients as early as possible to develop individualized treatment plans to improve treatment efficiency.
Copper is an essential cofactor for many biological processes occurring in the organism, but its concentration above a certain threshold can be toxic and induce cell death.Tsvetkov et al.[2]found that excessive intracellular copper triggers aggregation of mitochondrial lipoylated proteins and destabilization of Fe-S cluster proteins, leading to proteotoxic stress, inhibition of mitochondrial respiratory regulation and induction of cell death.The effect is different from that of previous regulatory cell deaths.Unlike previous regulatory cell deaths (e.g., apoptosis, iron apoptosis, and necrosis), this novel cell death is termed “cuproptosis”.In addition,it has been shown that cells dependent on mitochondrial respiration are 1,000 times more sensitive to copper ions than cells dependent on glycolysis, and the cuproptosis mechanism may be more sensitive to cells sensitive to mitochondrial respiration.Another study found that mitochondrial respiration function has an important role in the progression of GC, inhibition of mitochondrial respiration by knockdown of TIMMDC1 (membrane protein located in channel 4 of the inner mitochondrial membrane) significantly inhibited the proliferation and metastasis of GC cells, and mitochondrial respiration function may have an important role in the development of GC[3].Meanwhile, it has been demonstrated that disulfiram(DSF)/copper complexes can inhibit the proliferation and metastasis of GC cells through multiple pathways[4,5], and copper chelators are also expected to become therapeutic agents that break through the bottlenecks of drug resistance in a variety of cancers such as breast cancer, hepatocellular carcinoma, lung cancer and melanoma[6-9].Therefore, an insight into the role of cuproptosis mechanisms in GC is of great value.
Long noncoding RNAs (lncRNA)s are RNA transcripts that are greater than 200 nucleotides in length but do not encode proteins[10].LncRNAs regulate the expression of 70% of all human genes and can interact with DNA, RNAs, and proteins to exert either enhancement or inhibition[11].Although clinical applications of exome sequencing have identified many protein-coding mutations suitable for targeted therapies, the coding genome represents less than 2% of all gene sequences[11].There is growing evidence that LncRNAs are more precisely regulated than mRNAs in specific cell types, and that mutations in LncRNAs are equally important drivers of cancer phenotypes[12].For example, certain LncRNAs promote gallbladder cancer cell proliferation[13], inhibit ovarian cancer metastasis[14],and enhance breast cancer chemoresistance[15].LncRNA ABL is significantly overexpressed in GC cells, and its inhibition of caspase enzyme activation by binding to APAF1 promotes GC cell valueaddition[16].Therefore, LncRNAs hold significant promise for the study of tumor pathogenesis and therapeutic strategies.
Notably, the therapeutic, prognostic, and immune function assessment value of cuproptosis-related lncRNAs (CRLs) in GC has not yet been systematically evaluated, and the mechanism of cuproptosis in GC remains to be further elucidated.In addition,traditional Chinese medicine (TCM), as a natural product, is of great value for the development of tumor therapeutic drugs, and its active ingredients not only have important driving roles in several cell death pathways, such as iron death[17], cellular pyroptosis[18], and apoptosis[19], but also have the advantages of safety and economy.Therefore, in this study, we used cuproptosis as an entry point to screen and evaluate the prognostic value of CRLs for GC, and explored the relationship between CRLs and immune cell infiltration,immune function, and chemotherapeutic drug sensitivity, as well as predicted traditional Chinese medicines with potential modulatory effects on CRGs, with the aim of providing a reference basis for the treatment, prognosis, and development of new drugs for GC.
2.Materials and Methods
As this study was a re-analysis of previously published data, no additional ethical approval was required.
2.1 Data sources
We accessed clinical and transcriptomic data from The Cancer Genome Atlas (TCGA) database (https://portal.gdc.cancer.gov/projects/TCGA-STAD) for 443 GC patients.A Perl script was applied to obtain the normalized gene expression matrix for further analysis.We formalized Ensembl IDs into gene symbols and processed the data by log2.The perl script was then applied to distinguish lncRNA from mRNA by the annotation file of GENCODE v40 (https://www.gencodegenes.org/).
2.2 Acquisition of CRLs
From previous studies[2,20,21], 16 CRGs (NLRP3, ATP7B, ATP7A,SLC31A1, FDX1, LIAS, LIPT1, LIPT2, DLAT, PDHA1, MTF1, GLS,CDKN2A, DBT, GCSH, DLST) were identified, and the extraction of expression data of CRGs in 443 GCs.The expression data of CRGs and LncRNAs were analyzed by Pearson correlation analysis to obtain CRLs, and the filtering conditions were |Pearson correlation coefficient| > 0.4, P < 0.001.Pearson correlation coefficient > 0 represents positive regulation, and if it < 0, it is negative regulation.Sankey plots of CRGs and CRLs were drawn using the R “ggplot2”,“ggalluvial” and “dplyr” packages.
2.3 Construction and Validation of Prognostic signature
GC patients (All) were randomized into the Training (Train)and Validation (Vali) groups according to 7:3.CRLs significantly associated with prognosis (P < 0.05) were screened by univariate Cox regression in the Train group, and Hazard Rates (HR) were used to identify CRLs as risk factors (HR > 1) or protective factors(HR < 1).The CRLs were subsequently screened again by applying the Least Absolute Shrinkage and Selection Operator (LASSO)regression.To avoid overfitting, a 10-fold cross-validation method was used to select the optimal penalty parameters.The CRLs identified by the LASSO regression were finally included in the multivariate Cox proportional risk regression, and the risk model was constructed by calculating the risk score for each CRL.The risk score was calculated as follows:
N indicates the number of CRLs in the risk model, the relevant regression coefficient of CRLs in the multivariate Cox regression analysis, and the expression level of CRLs.
Based on the median risk score, GC patients in the Train and Vali groups were categorized into low-risk and high-risk groups,respectively.The accuracy of the risk model in predicting GC survival was assessed by comparing the Overall Survival (OS)and Progression Free Survival (PFS) of the high- and low-risk groups through Kaplan-Meier (KM) curves.Receiver operating characteristic (ROC) curves were applied to predict patient survival at 1, 3, and 5 years to validate the accuracy of the risk model.
2.4 Construction of the nomogram
To improve the accuracy of predicting GC survival, R “survival”and “RMS” packages were applied to construct nomogram combining clinical characteristics, and the accuracy of the nomogram was evaluated by ROC curve, calibration curve, and C-index.Subsequently, univariate and multivariate Cox regression analyses were performed for age, sex, Stage, Grade, and risk score to assess whether risk score could be an independent prognostic factor for GC.
2.5 Clinical subgroup analysis and principal component analysis
The GC patients were categorized into early (stage I-II) and advanced (stage III-IV), and the early and advanced groups of GC patients were divided into high- and low-risk groups, respectively,based on the median risk scores, and the differences in survival rates between the high- and low-risk groups were compared using KM survival curves in order to test whether the risk model could assess the prognosis independently of the clinical staging.The ability to group CRLs in the model was verified by Principal Component Analysis (PCA).
2.6 Functional enrichment analysis
To investigate the relationship between risk scores and biological processes, GC patients were categorized into high-and low-risk groups based on median risk scores, and differential genes between groups were identified using the R “limma” package, with the screening criteria of | log2 (fold change) | > 1 and FDR < 0.05.Gene ontology enrichment analysis of differential genes based on the“clusterProfiler” package (adjusted P < 0.05).
2.7 Analysis of immune function, mutated genes and drug sensitivity
Patients with GC were categorized into high- and low-risk groups based on the median risk score.Based on single-sample gene set enrichment analysis (ssGSEA), the infiltration scores of 16 immune cells, 13 immune-related pathways, and the differences in the expression of immune checkpoint inhibitory genes were compared between the high- and low-risk groups to determine the relationship between the risk scores and immune function.To evaluate the relationship between the risk score and mutated genes, the mutation frequency of cancer-related genes in the high- and low-risk groups was visualized using the R “maftools” package based on the tumor mutational burden (TMB) data of GC.Finally, the 50% inhibitory concentration (IC50) of chemotherapy/targeted drugs was evaluated using the “pRophetic” algorithm to determine the value of risk scores in predicting drug sensitivity.Differences between groups were assessed using the Wilcoxon signed-rank test.
2.8 Prediction of Traditional Chinese Medicine for CRGs
CRGs were loaded into Coremine database (https: //coremine.com/medical/#search) to map out the Chinese medicines with potential regulatory effects, and the screening condition was Significance< 0.01.The distribution of medicinal properties, medicinal flavor,meridian tropism and efficacy of Chinese herbal medicines is visualized through the ancient and modern cloud medical case platform (https://www.yiankb.com/).
2.9 Statistical methods
CRLs were determined using Pearson correlation analysis.In the Train group, risk models were constructed by univariate Cox,Lasso, and multivariate Cox regression.Differences in clinical characteristics between the Train and Vali groups were performed by chi-square tests.The accuracy of the risk model was assessed by timeROC curve and C-index.Differences in immune infiltration and chemotherapeutic drug sensitivity between groups were assessed by Wilcoxon signed rank test with R language version 4.2.2.
3.Results
3.1 Acquisition of CRLs
The clinical characteristics of the 443 GCs in the TCGA-STAD cohort in this study are shown in Table 1.
Transcriptomic data were obtained for only 407 GCs from the TCGA-STAD cohort, including 19,939 mRNAs and 16,876 lncRNAs.429 CRLs were screened by Pearson correlation analysis,as shown in Figure 1.
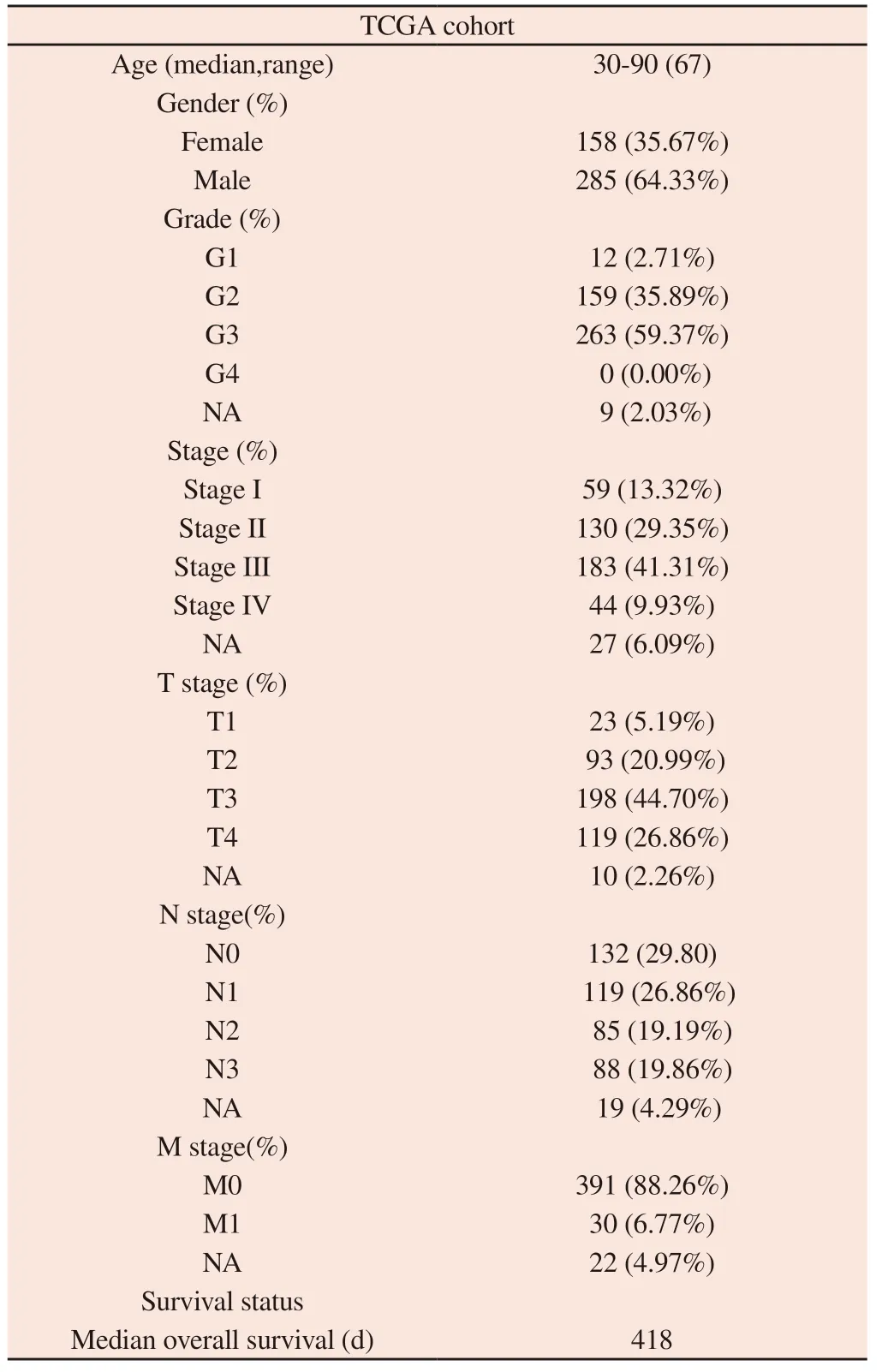
Tab 1 Clinical characteristics of 443 gastric cancer
3.2 Construction and validation of risk model
After deleting GC patients with missing clinical data, a total of 371 patients were randomly assigned to the Train group (N = 260) and the Vali group (N = 111), with no difference in clinical characteristics between the two groups, which were comparable, as shown in Table 2.In the Train group, 27 CRLs associated with survival prognosis were obtained by univariate Cox analysis (Figure 2A), of which 4 were protective genes (HR<1) and 23 were risk genes (HR>1).Subsequently, based on LASSO regression and multivariate Cox regression analyses (Figures 2B and C), 7 CRLs (AP001107.9,VCAN-AS1, AC016394.2, LINC02675, AC100814.1, HAGLR,LINC01094) were finally established to be included in the risk model.The correlations between the seven CRLs and CRGs in the risk model are shown in Figure 2D.
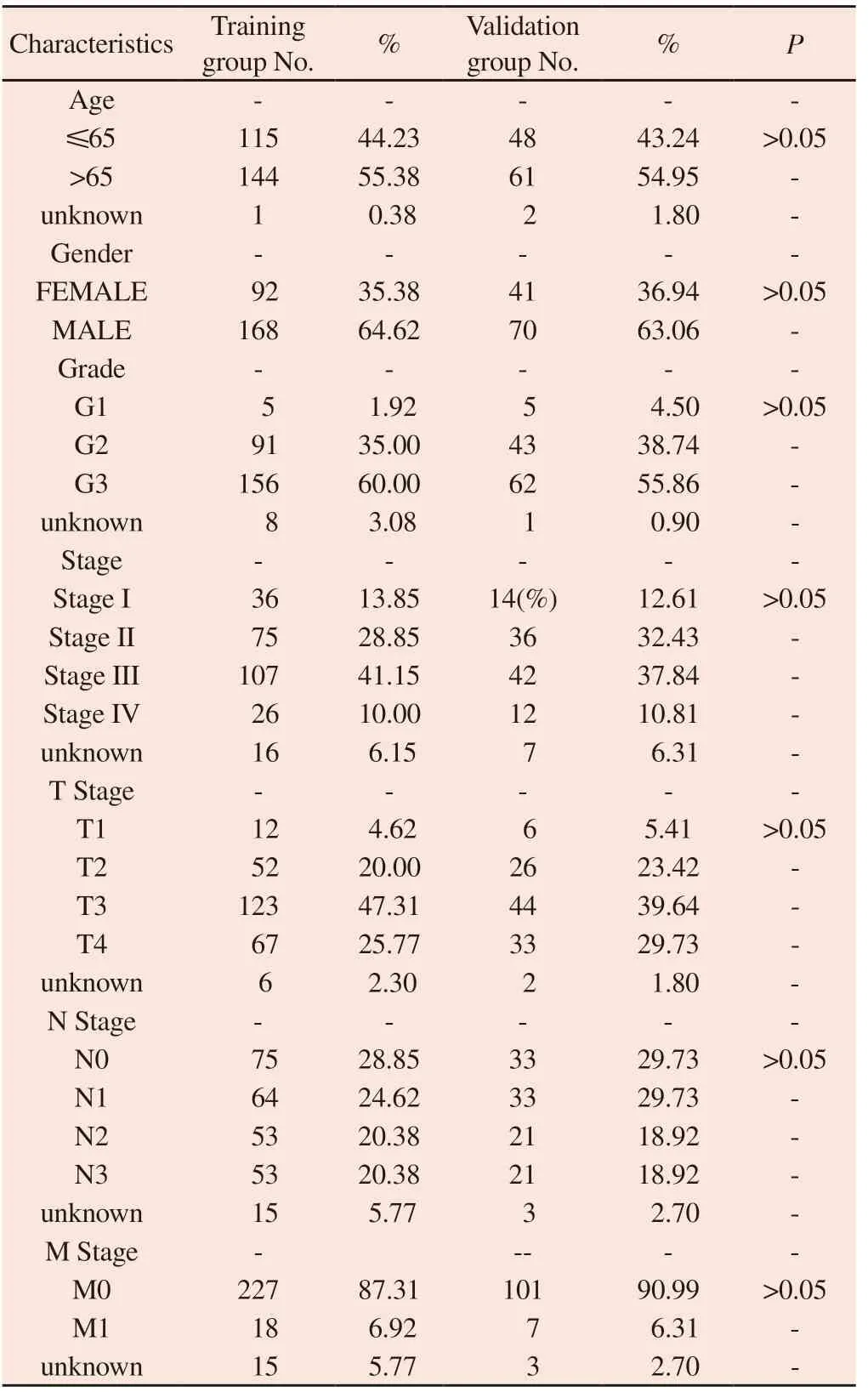
Tab 2 The comparison of clinical characteristics of GC patients between Train and Vali groups
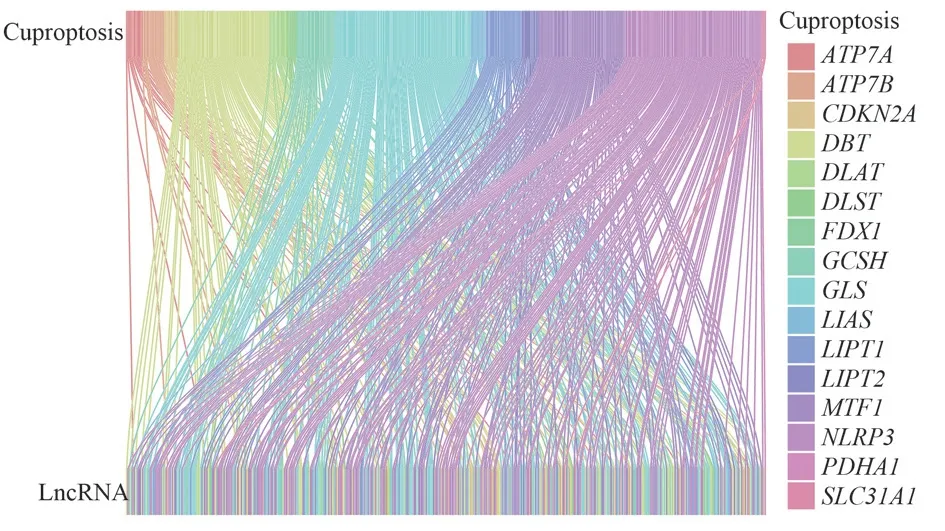
Fig 1 Sankey diagram of the association between CRGs and LncRNAs
We constructed a prognostic signature by calculating the risk scores of 7 CRLs.Risk scores were calculated from each lncRNA expression and correlation regression coefficient.Risk scores were computed by the following formula: Risk scores = (-0.215× expression level of AP001107.9) + (0.688 × expression level of VCAN-AS1) + (-0.416 × expression level of AC016394.2) + (0.459× expression level of LINC02675) + (0.253 × expression level of AC100814.1) + (0.196 × HAGLR expression level) + (0.583 ×LINC01094 expression level).GC patients in the Train (Figure 3A)and Vali (Figure 3B) groups were categorized into high-risk and low-risk groups, respectively, based on the median risk scores.The number of deaths in the high-risk group was significantly higher than that in the low-risk group in both groups (Figures 3C and 3D).There were differences in the expression of CRLs between the highand low-risk groups in the Train group (Figure 3E) and the Vali group (Figure 3F), and the expression of two protective lncRNAs(AP001107.9 and AC016394.2) was increased in the low-risk group,and five risk lncRNAs (VCAN-AS1, LINC02675, AC100814.1,HAGLR, LINC01094) were increased in the high-risk group.Survival analysis showed that OS and PFS were significantly worse in the high-risk group than in the low-risk group (Figures 3G-3J, P< 0.05).The risk model predicted AUCs of 0.720, 0.682, and 0.711 for 1-, 3-, and 5-year survival in the Train group (Figures 3K), and AUCs of 0.713, 0.620, and 0.676 for 1-, 3-, and 5-year survival in the All group (Figure 3M).
3.3 Independent prognosis analysis of prognostic signature and the construction of the nomogram
The independent prognostic efficacy of the risk model was checked by univariate Cox regression analysis and multivariate Cox regression analysis.The results showed that the risk model could assess the prognosis of GC independently of other clinical characteristics (P < 0.001) (Figures 4A and 4B).A nomogram was further constructed by combining clinical characteristics and risk scores.As shown, if a patient had a total score of 310, his probability of survival in the next 1, 3, and 5 years would be 75%, 39%, and 24.9%, respectively (Figure 4C).The calibration curves at 1, 3, and 5 years demonstrated the accurate prognostic power of the nomogram(Figure 4D).The risk model was more accurate than the prognostic efficacy of age, sex, TNM staging, and Grade classification (Figure 4E).In the Train group (Figure 4F) and Vali group (Figure 4G), OS prediction of GC patients based on nomogram was significantly higher than other predictors.Therefore, the risk model combining clinical characteristics can better predict the survival prognosis of GC patients.
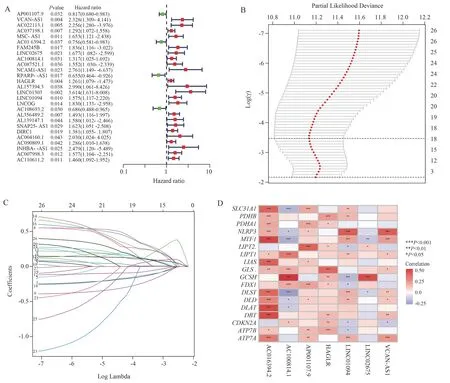
Fig 2 Construction of the risk signature

Fig 3 Prognostic efficacy of the risk signature
3.4 Clinical subgroup analysis and PCA analysis
After grouping GC patients by Stage, the survival rate of GC in the high-risk group was significantly lower than that of the lowrisk group in the subgroups of Stage I-II and Stage III-IV (P < 0.05)(Figures 5A and 5B), suggesting that the ability of the risk model to assess prognosis can be independent of clinical staging.In addition,PCA analysis showed that expression based on all genes (Figure 5C), CRGs (Figure 5D), or CRLs (Figure 5E) could not significantly differentiate the high-risk group from the low-risk group.However,based on the expression of the seven CRLs in the risk model, GC patients could be significantly differentiated into high-risk and lowrisk groups (Figure 5F).Therefore, the risk model can provide better prognostic risk assessment for GC.
3.5 GO enrichment analysis of differential genes
A total of 304 differential genes were obtained, and GO enrichment analysis of the differential genes showed that biological process(BP), cellular component (CC) and molecular function (MF) were significantly enriched in the extracellular matrix (ECM) (Figure 6).
3.6 Comprehensive immune infiltration analysis for risk modeling
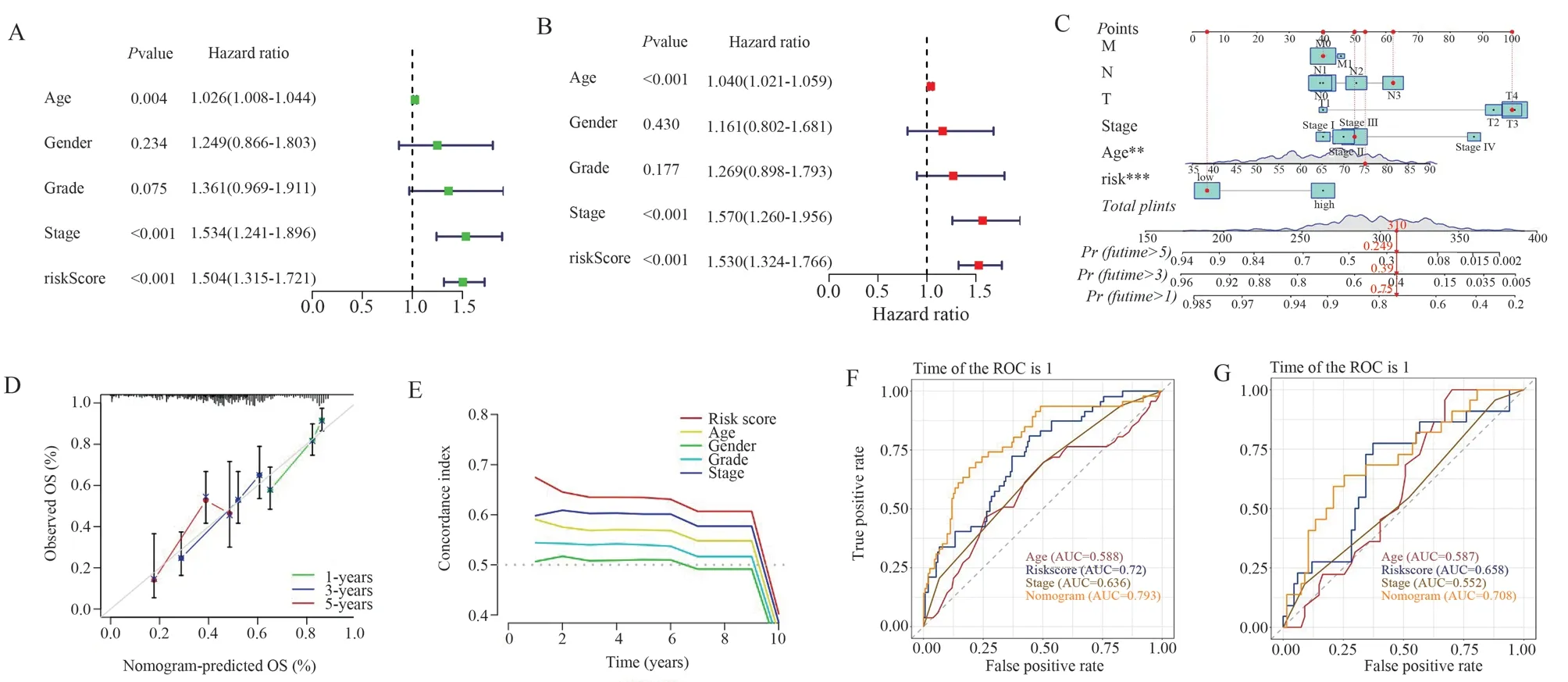
Fig 4 Independent prognostic capability of the risk signature and construction of nomogram
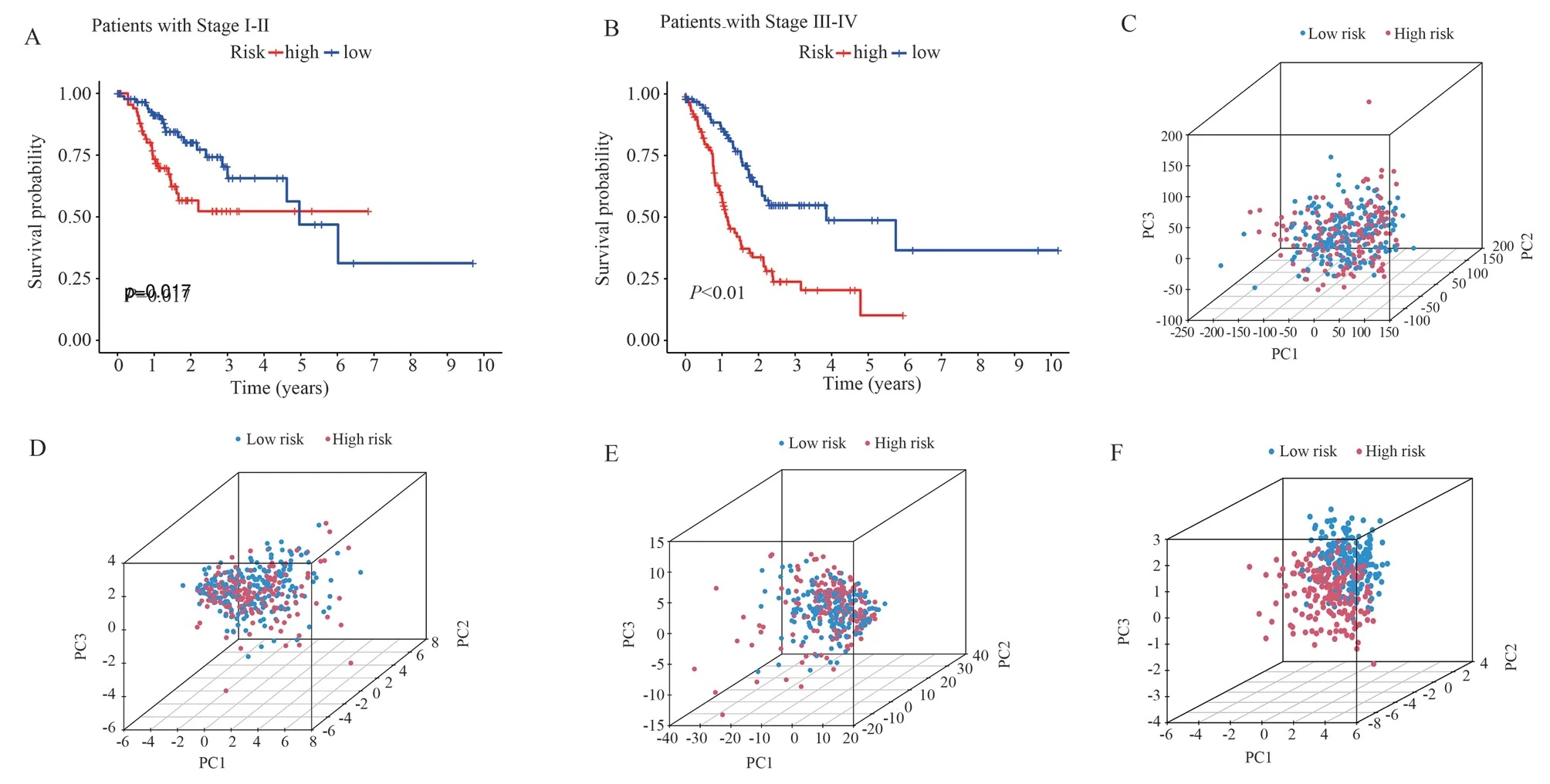
Fig 5 Survival curves for clinical subgroup types and PCA analysis
The ssGSEA analysis showed that the scores of antibody-drug couplers (aDCs), macrophages (Macrophages), neutrophils, plasma cell-like dendritic cells (pDCs), T helper cells, tumor-infiltrating lymphocytes (TILs), and regulatory T cells (Tregs) in the highrisk group were significantly higher than those of the low-risk group (P < 0.05) (Figure 7A).Differential analysis of immune function showed that antigen-presenting cell costimulation (APC costimulation), parainflammation, and chemokine receptor (CCR)were significantly upregulated in the high-risk group (P < 0.05)(Figure 7B).In addition, 22 immune checkpoint genes were significantly overexpressed in the high-risk group (P < 0.05) (Figure 7C).In conclusion, the immune cell score, immune function score,and expression of immune checkpoint genes were higher in the highrisk group than in the low-risk group, and the risk model can be used to predict the immune status of GC patients.
3.7 Analysis of cancer-related gene mutations and drug sensitivity
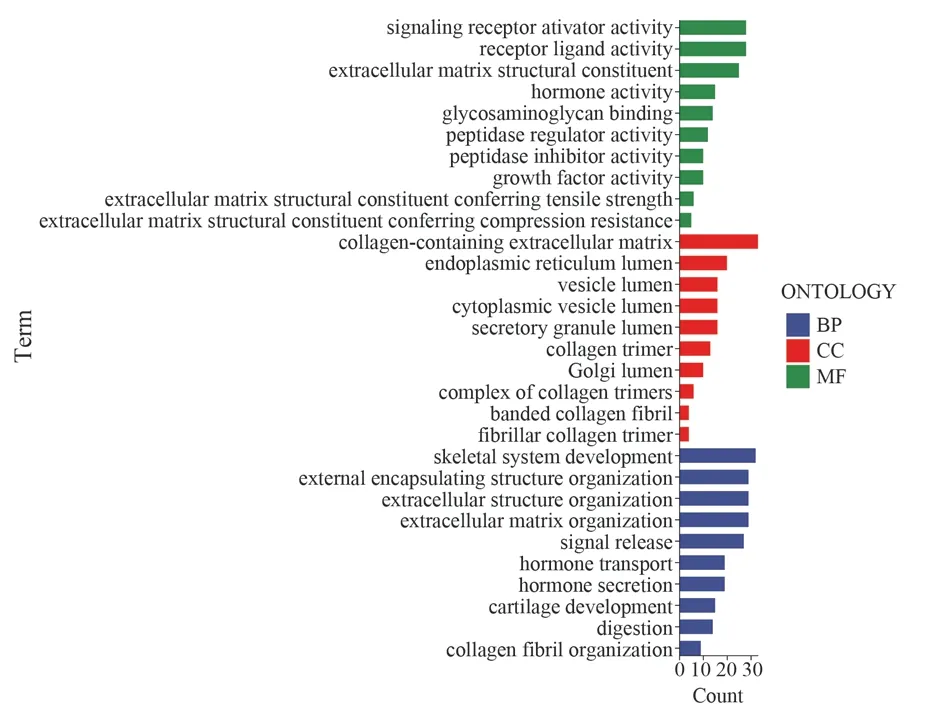
Fig 6 GO enrichment analysis of differential genes
The top 20 mutated genes in the high- and low-risk groups are shown in Figure 8, and the top five genes with the highest mutation frequencies in the high-risk group (Figure 8A) were the TNN (47%),TP53 (46%), MUC16 (24%), CSMD3 (24%), and LRP1B (23%)genes.TNN (53%), TP53 (38%), MUC16 (36%), ARID1A (32%)and LRP1B (30%) were the top five genes with the highest mutation frequency in the low-risk group (Figure 8B).Only the oncogene TP53 had a higher mutation rate in the high-risk group (46% versus 38%), whereas other cancer-related genes in the low-risk group had relatively high mutation rates.
3.8 Prediction of Traditional Chinese Medicine for CRGs
A total of 58 Chinese medicines mapped by 13 CRGs were obtained, as shown in Table 3, and the above Chinese medicines had potential regulatory effects on CRGs.The medicinal properties of the Chinese medicines regulating CRGs were mainly cold (Figure 11A), the medicinal flavors were sweet, bitter, and flat (Figure 11B), and they were mainly attributed to the liver, spleen, and lung meridians (Figure 11C), and their efficacies were mainly clearing heat and removing toxins, activating blood circulation, and relieving pain (Figure 11D).
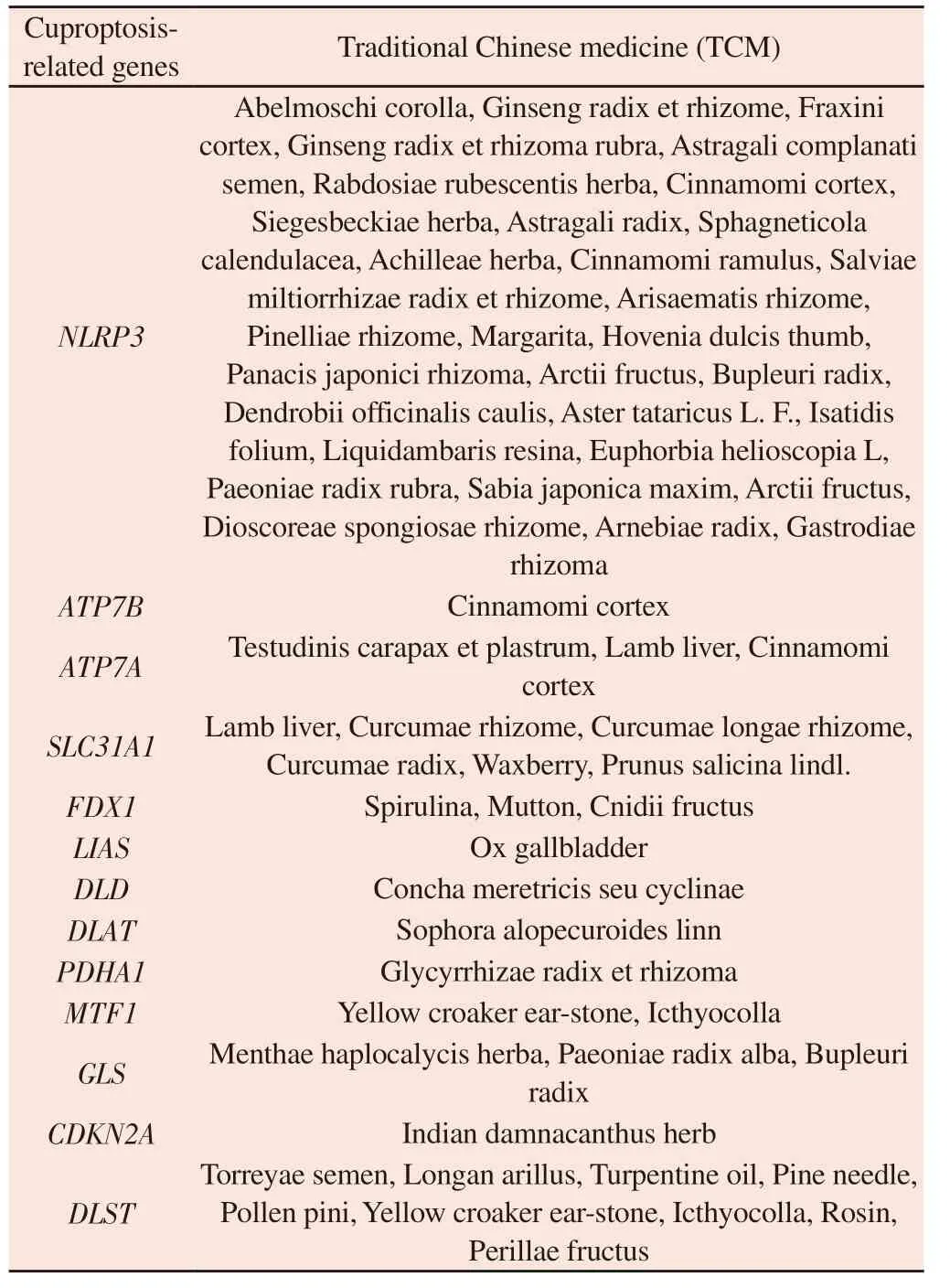
Tab 3 Prediction of cuproptosis related genes in Chinese medicine
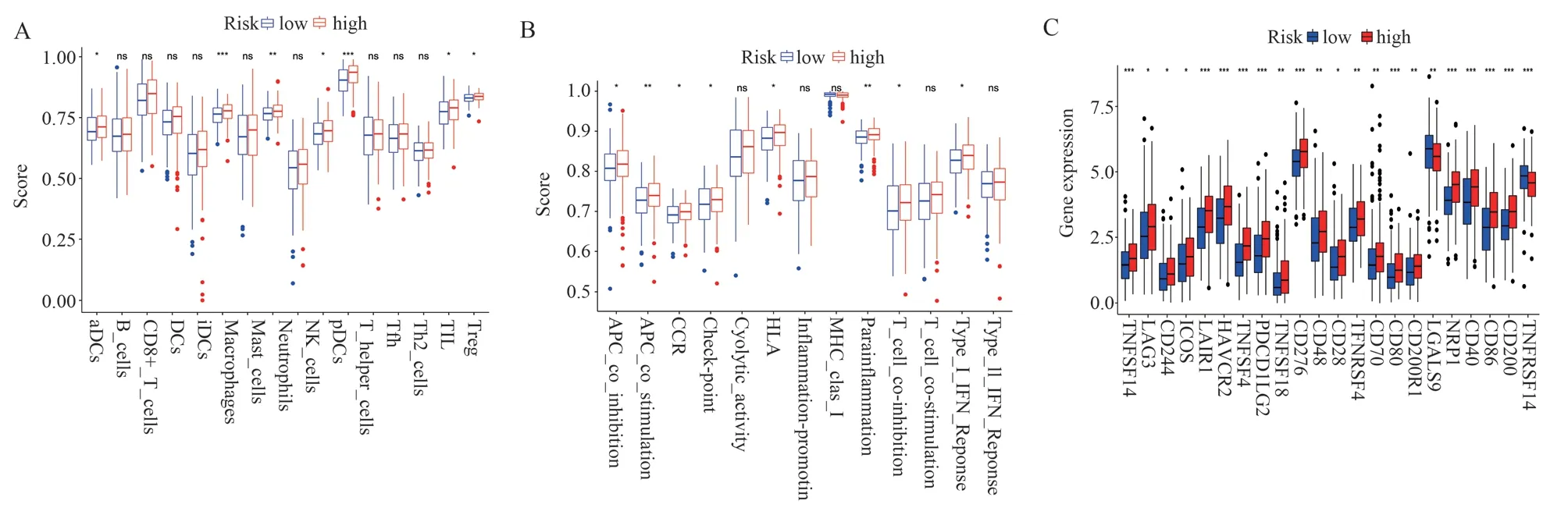
Fig 7 Relationship between risk signature and tumor immune microenvironment
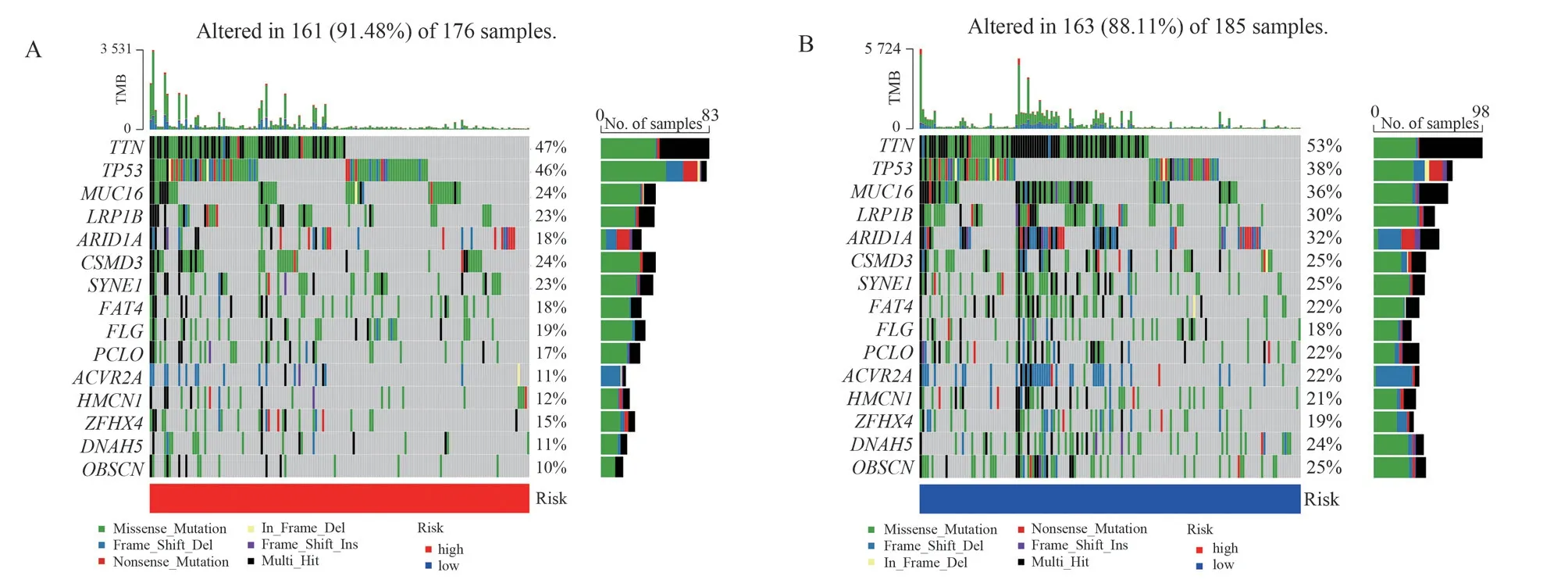
Fig 8 Waterfall plot of mutation frequencies in cancer-related genes
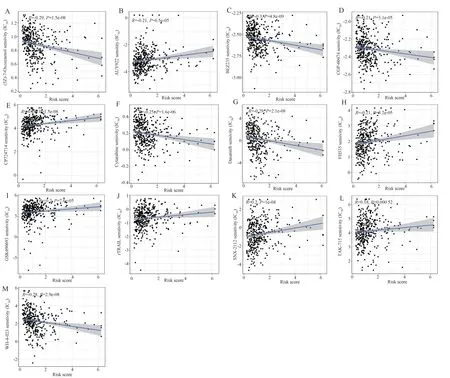
Fig 9 Scatter plot of correlation between IC50 values and risk scores for 13 drugs
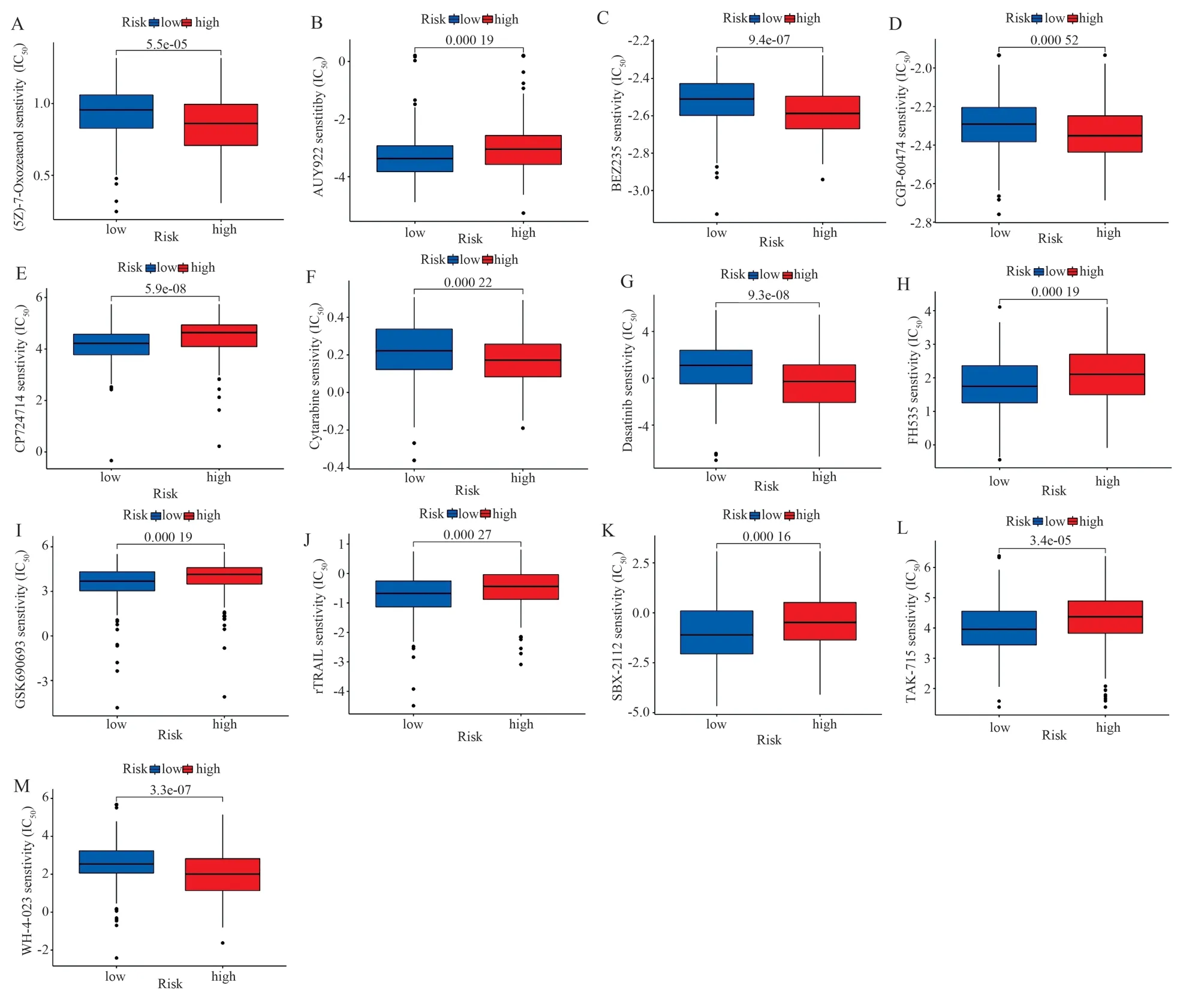
Fig 10 Boxplot of differences in sensitivity to 13 drugs between high and low risk groups
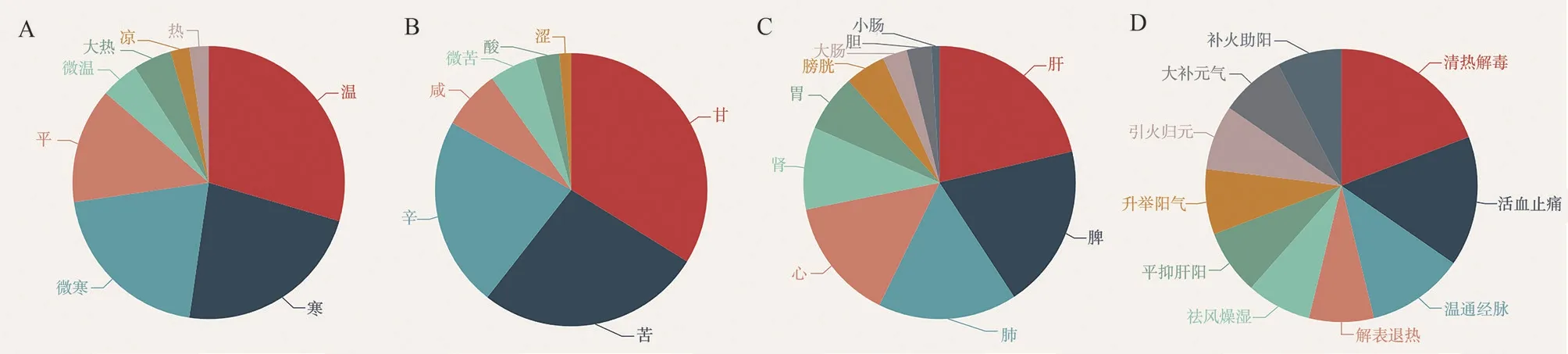
Fig 11 Medicinal properties, medicinal flavor, meridian tropism and efficacy distribution of Chinese medicines regulating CRGs
4.Discussion
Cell death can be categorized into accidental cell death (ACD) and regulated cell death (RCD), the latter of which is further divided into apoptotic and non-apoptotic RCD[22-24].For the past three decades,clinical oncology has been devoted to the development of therapeutic strategies to eliminate cancer cells by inducing cell death.Existing studies have shown that apoptosis resistance of cancer cells is the main cause of cancer treatment failure, and that elimination of cancer cells through the non-apoptotic RCD pathway is more effective than the apoptotic RCD pathway[22-25].Cuproptosis is a newly discovered mode of death, which is closely related to mitochondrial respiratory energy and belongs to a type of non-regulatory RCD[2].Based on the important role of mitochondrial respiratory regulation in GC cells[3],inducing apoptosis in GC cells by cuproptosis mechanism seems to be a promising research strategy, so it is valuable to explore the role of cuproptosis mechanism in GC in depth.
In this study, based on the clinical and transcriptomic data of 433 GC patients in the TCGA database, seven CRLs (AP001107.9,VCAN-AS1, AC016394.2, LINC02675, AC100814.1, HAGLR,and LINC01094) were finalized which were significantly associated with prognosis, and a risk model was constructed based on the expression level of each CRL and the associated regression coefficients to calculate the risk score to construct the risk model,and the risk model could better predict the survival prognosis of GC.The prognostic efficacy of some CRLs in the risk model for GC has been confirmed in previous studies.For example, Li et al.found that lncRNA VCAN-AS1 was significantly overexpressed in GC cells and significantly correlated with poor prognosis of GC,and negatively correlated with wild-type p53 expression[26].vCANAS1 significantly downregulated TP53 expression to promote the proliferation and metastasis of GC cells when combined with eIF4A3[26].lncRNA HAGLR had elevated levels of expression in both GC tissues and cells, and the expression level of HAGLR was increased in both GC tissues and cells.expression levels are elevated in both GC tissues and cells and correlate with poorer survival[27].It leads to 5-Fu resistance in GC cells by affecting lactate dehydrogenase a metabolism.Meanwhile, HAGLR plays an important role in the proliferation and metastasis of various tumors,including breast, liver, and esophageal cancers[28-30].Two other GC prognostic models based on autophagy-associated LncRNAs also incorporated HAGLR and LINC01094[31,32], suggesting that there may be a link between autophagy and cuproptosis, which was confirmed by Polishchuk et al.Knockdown of ATP7B activated autophagy-associated genes to protect the cells from copper-induced apoptosis[33].Therefore, the relationship between autophagy and cuproptosis deserves further in-depth study.However, the roles of AP001107.9, AC016394.2, LINC02675 and AC100814.1 in GC have not yet been reported, and the potential roles of the above LncRNs in GC still need to be further explored, and they are expected to be biomarkers for the assessment of GC treatment and prognosis.
The tumor immune microenvironment is important for the assessment of immunotherapeutic sensitivity and prognosis of GC.Comprehensive immune infiltration analysis in this study showed that there was abundant immune cell infiltration, such as tumor-infiltrating lymphocytes, macrophages, neutrophils, and regulatory T cells, in the high-risk group of GC patients.On the one hand, the poorer survival prognosis in the high-risk group may be related to the tumor immune microenvironment.The tumor immune microenvironment can be broadly classified into three types[34,35]: (1) immune-inflammatory: there is a large number of immune cells infiltrating inside the tumor; (2) immune-rejecting:although abundant immune cells are present, the immune cells are located in the interstitial matrix of the tumor cells, which cannot penetrate the interstitial matrix to reach the tumor parenchyma; and(3) immune-deserting: there is a lack of immune cell infiltration inside and around the tumor.Combined with the results of GO enrichment analysis, the differential genes between the two groups were significantly enriched in the ECM, so the patients in the highrisk group may have an immune-rejecting tumor microenvironment,in which the abundantly enriched immune cells are unable to penetrate the stroma to reach the tumor parenchyma to carry out effective immune clearance of the cancer cells, which leads to a poorer prognosis for their survival.On the other hand, elevated levels of tumor-infiltrating lymphocytes, macrophages, neutrophils,and regulatory T cells in the high-risk group of GCs predicted a poorer survival prognosis.Tumor-infiltrating lymphocytes include CD8+T cells, CD4+T cells, regulatory T cells, tumor-associated macrophages, and tumor-associated neutrophils[36].It has been found that high levels of macrophages, neutrophils and regulatory T cells in tumor patients are often indicative of a poorer survival prognosis[36].Thompson et al.found that GC patients with a poorer prognosis had higher levels of CD8+T cell infiltration[37].The present study similarly showed a higher level of CD8+T-cell infiltration in patients in the high-risk group, but the difference between the two groups was not statistically significant.As far as regulatory T cells are concerned, it has also been shown that their elevated peripheral blood levels predict poorer survival in GC patients[38].In addition,this study found a significant increase in neutrophil infiltration in patients in the high-risk group, which may be attributed to the high expression of the immunosuppressive molecule programmed death ligand 1 (PD-L1) by neutrophils, which allows tumors to evade immune surveillance and promotes tumor progression[39].In terms of immune function, the level of parvalidation was significantly higher in the high-risk group.Para-inflammation is a type of lowgrade inflammation that lies between typical inflammation and homeostasis, and is an adaptive response due to tissue stress or dysfunction.Aran et al.found that high levels of para-inflammation in tumors were positively correlated with p53 deletion, that parainflammation may be one of the main drivers that inactivate the p53 pathway, and that cancers with high levels of para-inflammation have a poor prognosis[40].The frequency of mutations in cancerrelated genes in this study similarly showed that the frequency of TP53 mutations was higher in the high-risk group (high parainflammatory levels) than in the low-risk group (46% vs.38%).In addition, 22 immune checkpoint genes showed high expression in the high-risk group, some of which have been identified as potential immunotherapeutic targets for GC.LAG3 is the third FDA-approved target for immune checkpoint inhibitors.Previous studies have also provided evidence that ICOS[41], CD28[42], and NRP1[43] are potential immunotherapeutic targets for GC, and that high-riskscoring GC may benefit more from treatment with the 22 immune checkpoint inhibitors mentioned above.
Among the 58 traditional Chinese medicines that regulate CRGs,the efficacy is based on clearing heat and removing toxins, activating blood circulation and relieving pain.Among them, FDX1[2] is the key gene regulating the cuproptosis mechanism, and the potential Chinese medicines acting on FDX1 are Spirulina and Cnidii fructus.Spirulina sun polysaccharide, the main component of Spirulina,has the functions of antioxidant damage and immune regulation,and can significantly inhibit the proliferation of GC cells[44,45].The main active ingredient of Cnidii fructus has anti-oxidative stress, antithrombotic and anticancer effects, and it can act on phosphatidylinositol 3-kinase/Akt protein kinase B pathway[46] and cystatinase-3-dependent apoptotic pathway[47] to induce apoptosis in GC cells.However, whether the above herbs can regulate the cuproptosis mechanism by modulating the FDX1 gene to induce cancer cell death has not been reported, and the regulatory effects of Spirulina and Cnidii fructus on the cuproptosis mechanism deserve to be explored in depth.
In summary, this study established a prognostic model of GC based on seven CRLs, which had excellent efficacy in prognosticating GC and could assess the immune function of GC patients, the expression of immune detection point genes, and their sensitivity to 13 chemotherapeutic agents, and the traditional Chinese medicines,such as Spirulina and Cnidii fructus, may have potential roles in regulating the mechanism of cuproptosis.
Author contributions: Zhangjun Yun and Yang Shen: article design,data analysis and manuscript writing; SuiCai Mi, Zhu Liu, Qiuyue Fan and Aiqi Liu: related literature collection and organization,data analysis; Miaojie Zhai: article formatting checking; Li Hou:providing overall ideas and revisions.
杂志排行

Journal of Hainan Medical College的其它文章
- Mechanism of AiTongXiao granule in the treatment of hepatocellular carcinoma based on network pharmacology and rat transplanted liver cancer model
- The effect and mechanism of stilbene glycosides on improving neuronal injury in Alzheimer's disease rats by regulating ASK/MKK7/JNK pathway
- Meta-analysis of the acupoint application therapy for stable chronic obstructive pulmonary disease
- Preparation of adhesive resveratrol micelles and determination of drug content
- BMSCs transplantation inhibits neuronal apoptosis after subarachnoid hemorrhage in rats through activation of AMPK/mTOR signaling pathway-mediated autophagy
- Research progress of necroptosis and ferroptosis in knee osteoarthritis