煤矸石精确产酸潜力评价方法对比研究
2024-01-18孙红福阴祥诚孙朗赵峰华朱孟浩范紫仪
孙红福,阴祥诚,孙朗,赵峰华,朱孟浩,范紫仪
中国矿业大学(北京)地球科学与测绘工程学院,北京 100083
煤炭开采过程中,由于黄铁矿等含硫化合物的氧化会产生大量酸性水,严重影响生态环境和煤炭安全高效开采[1]。酸性矿井水通常具有酸性高、硫酸根离子和重金属离子含量高等特点,大量的酸性矿井水一旦形成,其排放和治理成本巨大。矿山岩石产酸潜力评价能预测未来矿井水的水质特征,有助于合理预算矿山酸性水治理成本,有效避免重大酸性水污染事故[2-3]。酸性水预测的核心工作就是准确计算围岩和矸石的产酸潜力。传统产酸潜力的评价主要是基于岩石全硫含量计算的最大产酸潜力(Maximum Potential Acidity,MPA)。但矸石中不仅含有硫化铁硫,还有其他硫化物和硫酸盐。因此,有必要测定矸石中产酸的硫化物硫和硫酸盐硫含量。
煤矸石中总硫的测定可参考国标[4-5]。各形态硫含量则不能直接使用国标测定。如测定煤中硫化铁硫以硝酸提取的铁为基准,存在引入多种误差的可能[6-7]。早期测定矿石中硫化物硫和硫酸盐硫含量,主要采用550 ℃下热解、铬还原硫法和多种选择性湿法提取等方法[8-9]。550 ℃下热解常用于测定硫化物硫含量,但会受到易挥发的有机硫和硫酸盐干扰。铬还原硫法对价态小于+6 价的无机硫具有选择性,但只能得到还原性硫化物的总量[10]。选择性湿法提取的溶剂包括盐酸、硝酸、草酸铵等。以丙酮、热盐酸为主的逐级萃取可提取单质硫、酸溶性硫酸盐和酸挥发性的硫化物,但对其他含硫矿物无明显去除作用[11]。硝酸消化法会溶解非硫化物铁,测硫化物硫含量误差较大[12]。草酸铵酸浸法可完全提取氧化铁和硫酸盐矿物[13]。之后,发展出了结合上述方法的分步提取程序,针对煤炭废弃物中的含硫矿物种类测定较成熟的有ACARP C15034 协议、三步连续提取等[14-15]。三步连续提取法结合热解法和湿法提取,能有效区分并计算硫化物硫和硫酸盐硫含量。LI 等[15]用已知成分配比的含硫矿物组合验证了该方法的有效性。
本文通过识别矿石中常见产酸硫的种类,详细介绍含硫矿物产酸值的计算方法,采用三步连续提取法测定我国矸石中主要的产酸硫含量,并评估方法适用性。同时,还分析了精确产酸潜力(Precise Potential Acidity,PPA)法相比MPA 法对产酸预测精度的影响。
1 产酸潜力计算
1.1 产酸含硫矿物
矿山岩石产酸的来源,主要有硫化物矿物的氧化、酸性硫酸盐矿物的溶解、硫化物和硫酸盐矿物中金属的水解[16]。不同成分的含硫矿物,在产酸能力上有很大差异。因此,识别矸石中产酸硫的种类是准确预测产酸潜力的第一步。矸石中含硫矿物的常见类型有硫化物、硫酸盐()、有机硫和单质硫。硫化物和硫酸盐是主要的产酸含硫矿物;有机硫通常不产酸;单质硫含量低且产酸慢,可忽略。
硫化物包括单硫化物(S2-)、复硫化物(、AsS2-等)和硫盐(等)。根据产酸程度可分为:①产酸硫化物,包括黄铁矿(FeS2)、磁黄铁矿(Fe1-xS)、砷黄铁矿(FeAsS)等。②不产酸硫化物,包括闪锌矿(ZnS)、方铅矿(PbS)等。③部分产酸硫化物,包括黄铜矿(CuFeS2)、镍黄铁矿[(Fe,Ni)9S8]等。
硫酸盐矿物在煤矸石中含量不高,但也是产酸中不可忽视的部分。硫酸盐根据产酸程度分为:①产酸硫酸盐,包括泻利盐(MgSO4·7H2O)、黄钾铁矾[KFe3(SO4)2(OH)6]、水绿矾(FeSO4·7H2O)。②非产酸硫酸盐,包括石膏(CaSO4·2H2O)、芒硝(Na2SO4·10H2O)、重晶石(BaSO4)。
综上,可将煤矸石中产酸的含硫矿物分为产酸的硫化物和硫酸盐、部分产酸硫化物。
1.2 最大产酸潜力计算
MPA 由公式(1)计算。
式中,w(MPA)为样品的最大产酸(以H2SO4计)潜力,kg/t;w(St)为样品的全硫质量分数,% ;30.6为系数。
1.3 精确产酸潜力计算
1.3.1 黄铁矿产酸潜力计算
近中性条件下,黄铁矿完全氧化的产酸量计算基于反应式(2)和式(3),即1 mol 硫产生2 mol H+,1 mol 方解石中和2 mol H+。
黄铁矿产酸潜力(以H2SO4计)参考式(4)。
式中,w(PPApy)为样品中黄铁矿的产酸潜力,kg/t;w(Spy)为样品中黄铁矿硫的质量分数,% ;30.6 为系数。
只含铁元素的硫化物(磁黄铁矿、白铁矿)产酸的化学方程式和黄铁矿类似,与黄铁矿产酸潜力的计算公式相同。
1.3.2 其他硫化物和硫酸盐产酸潜力计算
近中性环境下,其他硫化物与氧气、水反应的产酸量,取决于反应终产物中自由离子的占比。
银、镉、钴、铜、镍、铅和锌的硫化物,如果金属离子发生完全的水解、置换、沉淀、络合,即没有自由的金属阳离子释放,则1 mol 硫产生2 mol H+,见式(5);如果金属离子为自由离子,则不产酸,如闪锌矿、方铅矿等在潮湿环境中直接氧化成二价金属离子和[17],见式(6);如果部分金属离子为自由离子,则1 mol 硫产生的H+小于2 mol。
砷、钼的硫化物,如果氧化形成的阴离子完全置换、沉淀、络合,即除外无其他自由的阴离子释放,则1 mol 硫产生2 mol H+;如果阴离子为自由离子,则1 mol 硫可产生3 mol 或4 mol 的H+,如反应式(7)和式(8)。
理论计算中,钼的终产物若是自由阴离子,则1 mol 硫产生3 mol H+;砷的终产物若是自由阴离子,假设产酸最大化,则1 mol 硫产生4 mol H+[16]。
当岩石中硫化物含量较少时,产酸硫酸盐的作用是不可忽略的。酸性硫酸盐(如水绿矾),1 mol硫酸盐硫产生2 mol H+,反应过程见式(9);含碱性阳离子的硫酸盐(如黄钾铁矾),1 mol 硫酸盐硫产生1.5 mol H+,反应过程参考式(10)。
综上,矿石的精确产酸潜力值可通过各个产酸硫化物、产酸硫酸盐的产酸潜力值依次相加,由公式(11)计算。
式中,w(PPAtotal)为样品的精确产酸潜力值,kg/t,以H2SO4计;wi(S)为样品中第i个产酸含硫矿物硫的质量分数,% ;αi为第i个产酸含硫矿物硫相对黄铁矿硫的产酸倍数;k为样品中的产酸含硫矿物数量。
矿山常见含硫矿物单位硫产酸系数和相对黄铁矿的产酸倍数见表1。

表1 常见含硫矿物每摩尔硫的产酸系数[18]Table 1 Acid production coefficient per mole of sulfur produced by common sulfur-containing minerals[18]
2 材料的制备与分析测试
2.1 样品采集与处理
采集了不同矿山的5 个煤矸石样品和5 个含硫矿石样品。具体编号如下:
所有样品研磨至小于200 目。独立取5 个矿石粉末样与石英粉末以质量分数5%、95% 的比例充分均匀混合[15],模拟矿区常见的以单一硫化物为主的矸石。
2.2 XRD 分析及全硫检测
使用X 射线衍射仪对10 个样品进行物相分析。每个样品各取5 g。仪器型号为Panalytical X’Pert PRO X 射线衍射仪,检测方法依据《矿物晶胞参数的测定——粉末X 射线衍射法:EJ/T 553—1991》进行。
全硫测定采用艾士卡法和红外光谱法。艾士卡法将5 类煤矸石样品和5 类矿物样品分别与艾士卡试剂混合灼烧,矿石中硫生成硫酸盐,然后使硫酸根离子生成硫酸钡沉淀,根据硫酸钡质量计算煤矸石中全硫含量[4]。
红外光谱法测定硫含量与国际标准ISO 19579—2006 接近,能快速准确获得检测数据[19],也用于测定残余物总硫。检测仪器是碳硫分析仪(Leco CS230)。
2.3 实验过程
2.3.1 实验试剂与仪器
试剂:30% 过氧化氢溶液,69% 硝酸溶液,4 mol/L 盐酸溶液,超纯水等。
仪器与材料:0.22 μm 聚四氟乙烯滤膜,坩埚,真空抽滤器,马弗炉,旋片式真空泵,BZF-30 真空干燥箱,ME204 电子分析天平,Agilent 730 ICPOES 光谱仪(检出限:Fe 0.1 μg/L、Mg 0.01 μg/L、Ca 0.01 μg/L、S 3 μg/L、K 0.3 μg/L、Cu 0.3 μg/L)。
2.3.2 实验步骤
实验采用三步连续提取法,流程如图1 所示。实验步骤[15]如下:

图1 三步连续提取法流程Fig.1 Three-step sequential extraction
(1) 萃取溶解度较高的硫酸盐。①称量(2±0.01)g 的样品,放入175 mL 塑料瓶中;②加入80 mL氩气吹扫过的水,制成1 ∶40 的悬浮液,在封闭瓶子前用氩气吹扫排出瓶内的空气;③塞紧瓶塞并在混合恒温箱中20 ℃下萃取3 min;④用0.2 μm 的特氟龙膜过滤溶液,得到纯净的提取物,并分成两份:一份加两滴30%的过氧化氢溶液测定酸度;另一份用两滴69%的硝酸酸化(至pH 值约为1),以防止出现氢氧化铁沉淀,通过ICP-OES 测定溶液中铁、镁和硫的含量,计算出水绿矾、泻利盐等可溶硫酸盐含量;⑤平行样用Leco CS230 测定水萃取后滤饼总硫。
(2) 除去黄铁矿等铁硫化物和部分铜硫化物。①用氩气吹扫水(4×10 mL)仔细清洗上一步实验的滤纸上的残余固体;②将滤饼置于干燥箱中120 ℃干燥2 h;③干燥后的滤饼置于坩埚中,在马弗炉中550 ℃焙烧1 h,去除黄铁矿;④平行样用Leco CS230 测定焙烧后剩余固体的总硫。
(3) 盐酸萃取剩余的铜硫化物、酸溶性的硫酸盐[如黄钾铁矾KFe3(SO4)2(OH)6],并测定最后剩余的硫。①等焙烧的滤饼冷却后,转移到萃取瓶中;加入4 mol/L 的盐酸80 mL 并静置萃取30 min;②用0.2 μm 特氟龙膜过滤悬浮液,稀释10 倍后用ICP 分析萃取液;③用氩气吹扫水洗净残余物;④将固体残留物转移到坩埚,在干燥箱中105 ℃干燥3 h;⑤用Leco CS230 测定残余总硫。
每批样品采用2 组平行样,分别测定水萃取后剩余物、焙烧后剩余物的总硫,计算得到焙烧过程的硫损失。该法分步提取含硫矿物,减少了各种含硫矿物的相互干扰。
3 结果和讨论
3.1 XRD 及硫含量测定结果
XRD 检测结果见表2。图2 为10 个样品的XRD 图谱。其中,样品MGS、HB、HN 未检出含硫矿物,可能含硫量较低或以非结晶态存在。
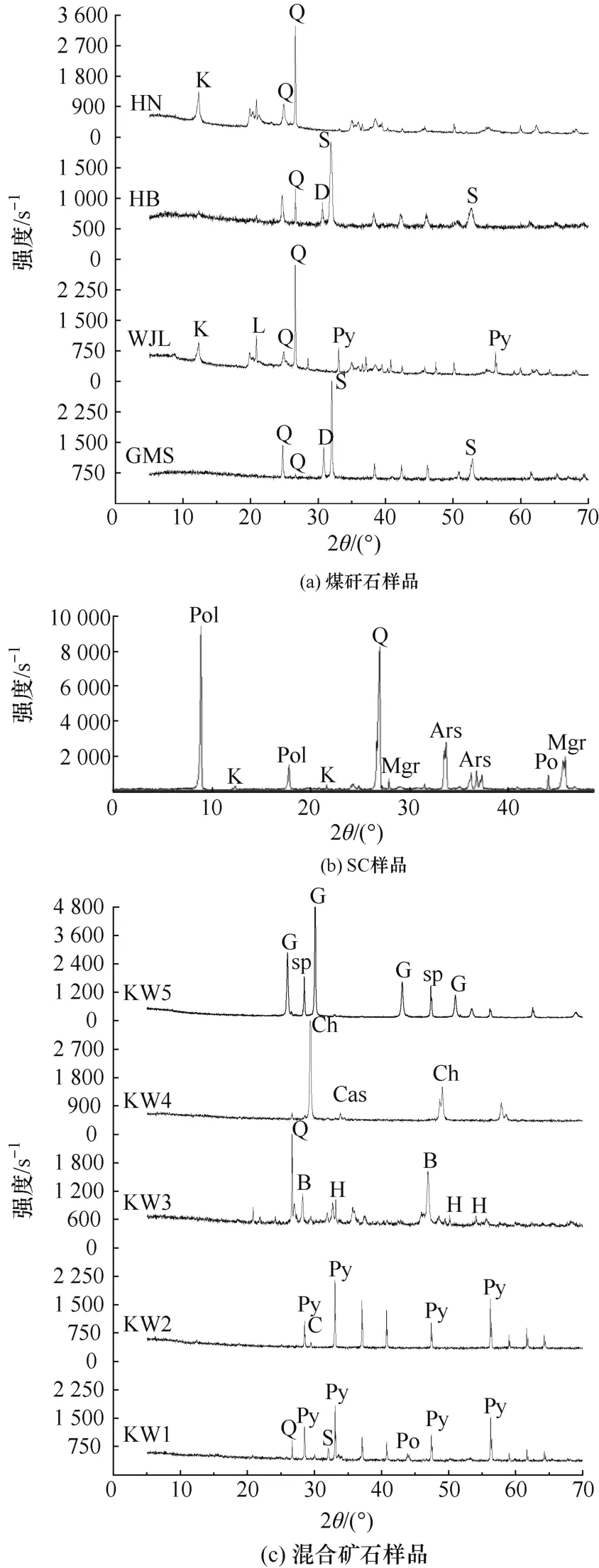
图2 10 个样品的XRD 图谱Fig.2 XRD patterns of ten samples

表2 XRD 检测结果Table 2 XRD test results of samples
艾士卡、红外光谱法的全硫检测及各步提取后残余物的总硫检测结果见表3。可以看出:样品WJL 水萃取后总硫测定中,总硫反而高于原岩;样品MGS、HB 焙烧后总硫测定中,总硫也高于原岩。分析原因,样品WJL 可能是水萃取步骤存在数据检测错误;样品MGS 和HB 焙烧后样品可能和组分有关,两者物相检测均含白云石或菱铁矿且未检出含硫矿物,可能是菱铁矿和白云石在焙烧过程中分解,导致样品质量降低;硫的质量分数低,焙烧中硫的损失对样品整体影响不大,导致样品中硫的质量分数相对增大。
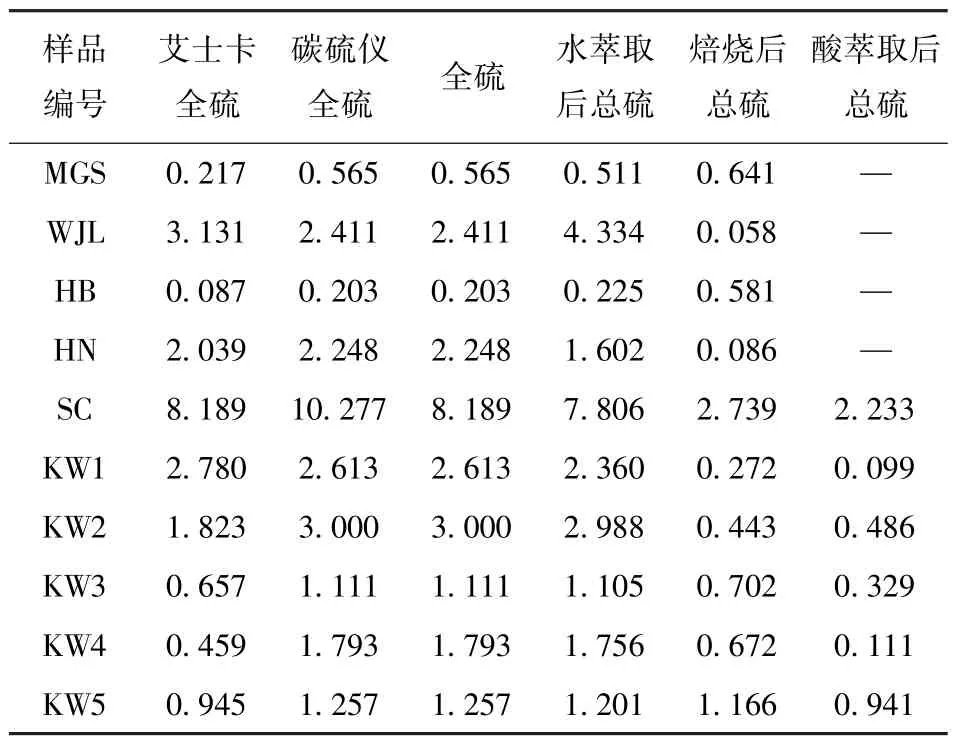
表3 硫测定结果占原样的质量分数Table 3 The ratio of sulfur determination results to the mass of the original sample%
从表3 还可以看出,两种方法测得的全硫结果中,SC、KW2、KW3、KW4 和KW5 样品差异较大。原因可能是:SC 含硫量高,超出碳硫分析仪5% 的准确检测上限,故应采用艾士卡法结果;KW2、KW3、KW4 和KW5 样品和艾士卡试剂未混合均匀,造成艾士卡法结果都低于碳硫分析仪结果,因此采用碳硫分析仪的全硫结果。HN 和KW1 和测残余物总硫的方法一致,也采用碳硫分析仪结果。
3.2 三步连续提取实验结果
ICP-OES 检测萃取液中元素含量见表4 和表5。各步骤中硫占原样的质量分数见表6。“各步骤硫的损失/计算总硫”的百分比见表7。表7 对应的直方图如图3 所示。
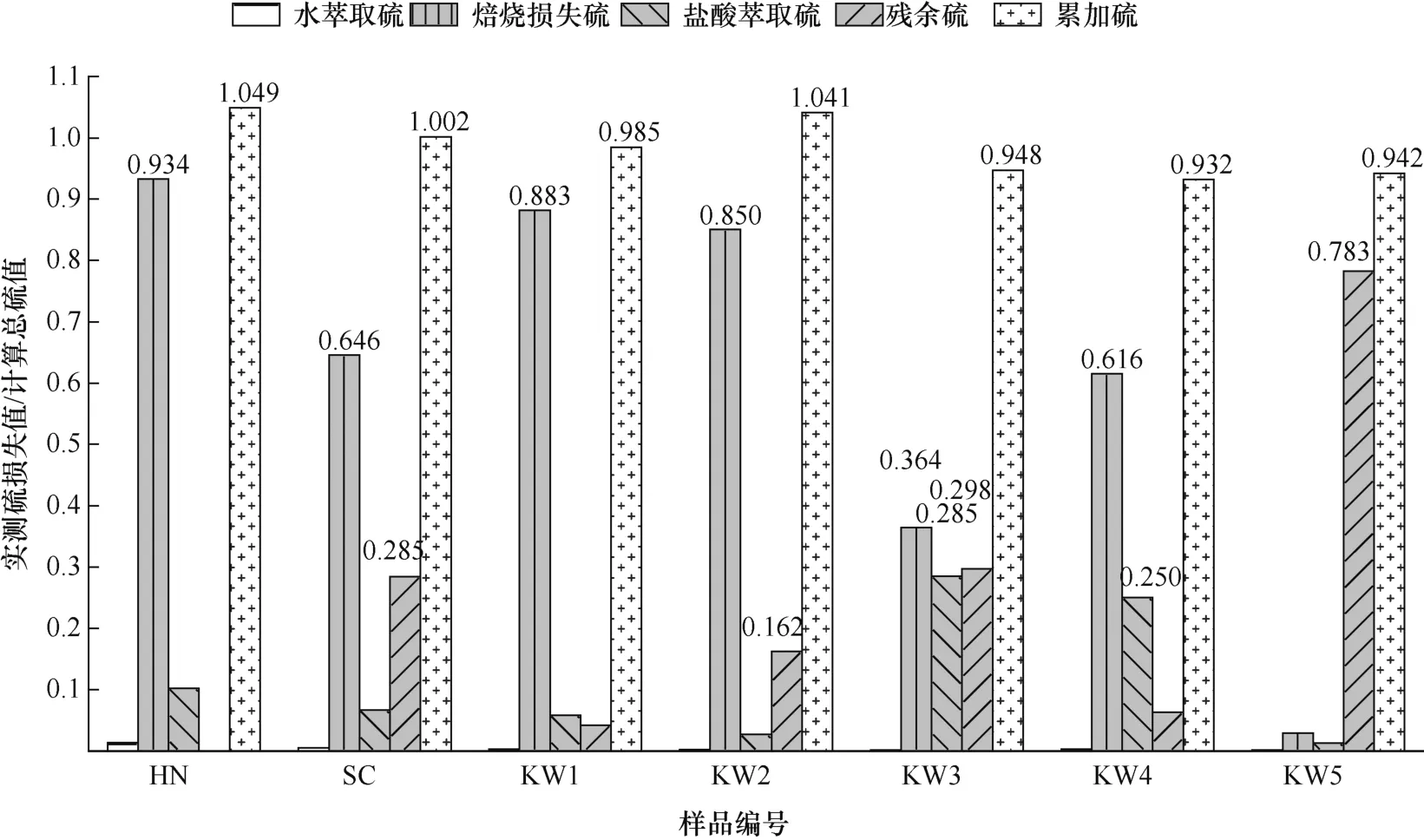
图3 各步骤“实测硫损失值/计算总硫值”Fig.3 Measured sulfur loss value/calculated total sulfur value in each step
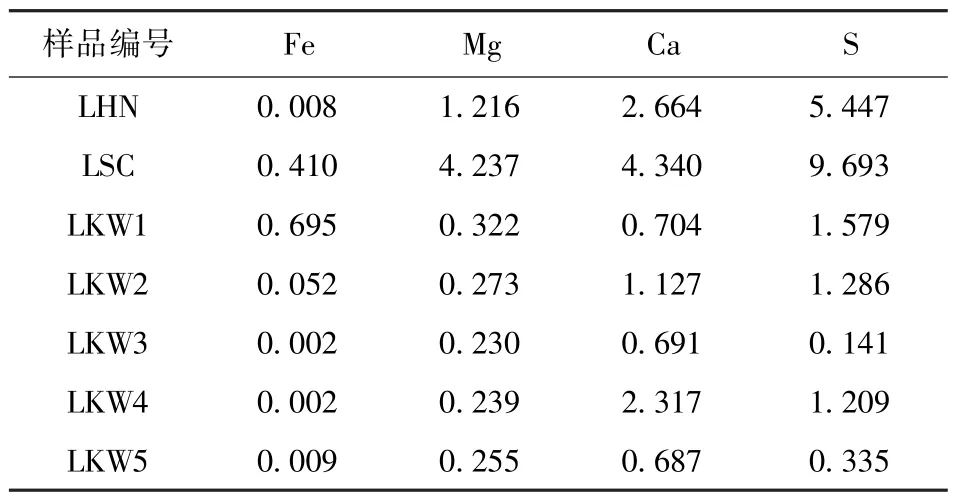
表4 水萃取液ICP 测定结果Table 4 ICP determination results of water extraction mg/L

表5 盐酸萃取液ICP 测定结果Table 5 ICP determination results of hydrochloric acid extractionmg/L

表6 各步骤中硫占原样的质量分数Table 6 The ratio of sulfur loss in each step to the mass of the original sample%
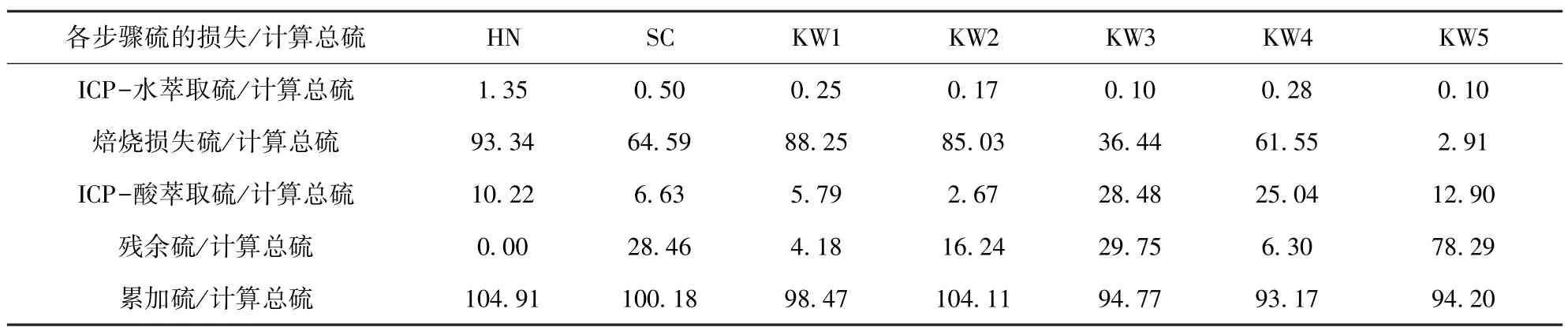
表7 各步骤“实测硫损失值/计算总硫值”Table 7 Measured sulfur loss value/calculated total sulfur value in each step%
表6 中,为排除水萃取过程平行样的硫损失过大的干扰,引入“计算总硫”(数值表示为“平行样-水萃取后总硫+ICP-水萃取硫”)取代实际全硫,作为硫损失的总硫参考值。ICP-水萃取硫、酸萃取硫是由ICP 结果经过式(12)换算得到。
式中,w(SICP)为ICP 萃取硫的质量分数,%;c(SICP)为ICP 提取物中的元素质量浓度,mg/L;m为提取的样品质量(默认是初始时的2 g),g;V为滤液体积,L;0.1 为从mg/g 转换为质量分数的换算系数。
焙烧损失硫为焙烧前后样品总硫含量的差值。残余硫是测定盐酸萃取后滤渣得到的。累加硫是ICP-水萃取硫、焙烧损失硫、ICP-酸萃取硫和残余硫的和。
表7 尾行计算了三步连续提取法在排除水萃取过程平行样干扰后的累加误差。误差小于±5%的是SC、HN、KW1 和KW2,结果满足误差要求。误差在5% ~10% 的是KW3、KW4 和KW5。
由于仪器和测定方法等原因,HN、KW3、KW4和KW5 样品的测定结果误差未能达到要求。HN 样品的总硫误差超过5%,原因可能是在测焙烧前滤渣的总硫时,样品水分烘干时间不够,而水能够吸收红外辐射,干扰红外线的测定精度,导致焙烧损失硫和总硫含量值偏高。KW3、KW4 和KW5 样品总硫含量偏低,原因可能是盐酸萃取步骤中,部分硫化物与盐酸反应,产生H2S 气体逸出,导致硫含量的损失;或者是由于在测定盐酸滤渣的残余硫时温度未达到1 300 ℃,没有将所有含硫矿物氧化成二氧化硫,导致残余硫测定值偏低。
3.3 实验各步骤中硫损失的讨论
实际水萃取的硫损失远大于ICP 结果,可能是平行样品的混合不均匀,导致测得水萃取后总硫过小。焙烧损失硫大多是以硫化铁硫为主的产酸硫化物,因为硫化铁硫在550 ℃焙烧下几乎完全去除(表8),而铜的硫化物只有部分热解,闪锌矿、方铅矿仅不到5% 热解。由于KW3、KW4 以铜的硫化物为主,KW5 以闪锌矿、方铅矿为主,导致KW3、KW4 和KW5 的焙烧损失硫百分比要明显小于其他样品。此外,焙烧1 h 后用冷盐酸萃取0.5 h 将有效去除剩余的铜硫化物,导致图3 中KW3 和KW4 的盐酸萃取硫高于其他样品。
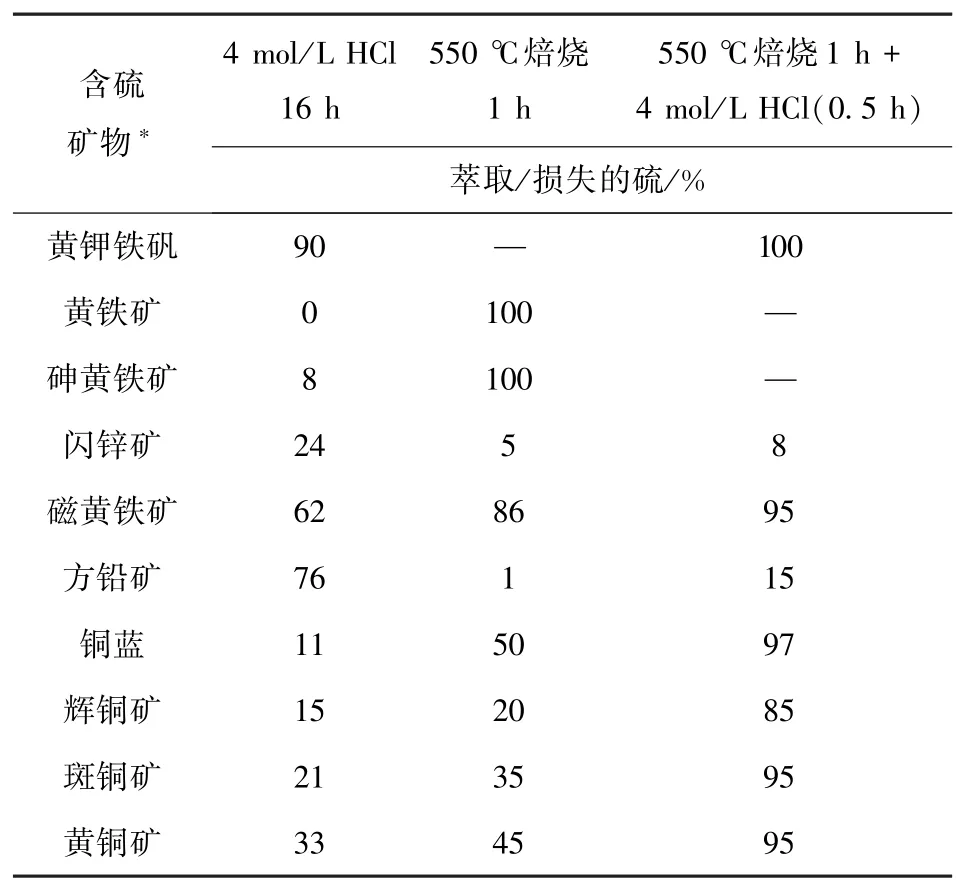
表8 三种提取方法对10 个单一成分含硫矿物的提取效果[15]Table 8 Extraction effects by the three extraction methods on 10 single-component sulfur-containing minerals[15]
盐酸滤渣中的残余硫可能是热稳定性强的有机硫,或是难以被焙烧和酸溶完全去除的硫化物和硫酸盐,如方铅矿、闪锌矿、重晶石等。SC、KW3 残余硫质量分数较高,可能是有机硫。KW5 残余硫质量分数最高与成分有关,方铅矿、闪锌矿经盐酸提取后仍有很多残留。
3.4 三步连续提取法评价
利用不同的矸石和矿石样品,初步验证了三步连续提取法提取主要产酸含硫矿物的有效性。该方法适用于成分以含铁、铜的硫化物为主的矿石。当矿石中其他硫化物成分增多时,会对产酸硫含量测定产生一定干扰。
该方法在以下方面有待改进:矸石样品550 ℃焙烧1 h 发生的各种矿物反应过程尚不完全清楚;部分热稳定性差的有机硫(硫醚、噻吩等)会在焙烧环节热解,增大焙烧损失硫[20-21];由于萃取液中阳离子存在多种矿物来源,以ICP 结果分析产酸矿物含量会受到一定干扰;矸石中的有机硫和未定形态硫对有机硫的定量,可考虑使用透射电子显微镜及能谱仪直接测定[22],对未定形态硫还需要更高精度的矿物学分析。
3.5 产酸潜力的精度分析
矿石的精确产酸潜力PPAtotal值的确定,是通过三步连续提取法测定各种目标产酸含硫矿物硫的含量后,再依据公式(11)计算得到的。
精确产酸潜力和最大产酸潜力的比较如图4所示。除SC 外,其余样品PPA 值均低于MPA 值,是由于全硫中除产酸的含硫矿物外,还包含不产酸的硫化物(闪锌矿)和硫酸盐(石膏)。当这些矿物含量较高时,用全硫计算的产酸潜力将大大高过实际情况。KW5 含硫成分主要为不产酸的闪锌矿和方铅矿,几乎不含产酸的硫化物,这造成了PPA 值接近于0,MPA 值是PPA 值的近200 倍。而样品SC 的PPA 值超过MPA 值,是因为含硫成分中主要为砷黄铁矿,而1 mol 砷黄铁矿完全产酸为1 mol 黄铁矿完全产酸的2 倍。其他样品MPA 值和PPA 值间的差异主要由全硫和产酸硫含量间的差距造成。与直接测全硫相比,依据式(11)计算PPA 值,提高了计算精度,更接近实际产酸潜力。
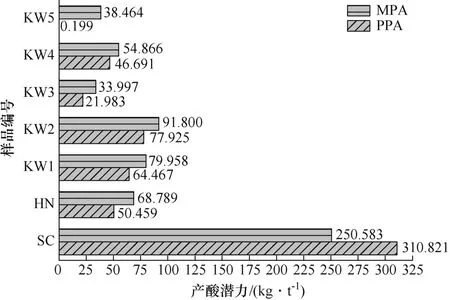
图4 样品的两种产酸潜力比较Fig.4 Comparison of acid production potential PPA and MPA of each sample
4 结 论
(1) 煤矸石的产酸潜力由产酸含硫矿物的种类和含量决定,产酸潜力值由各产酸硫占原样的质量百分含量乘以单位硫产酸值计算。
(2) 大部分样品用产酸硫含量预测的产酸值,均不同程度低于用全硫预测的产酸值。SC 样品中主要产酸硫化物(砷黄铁矿)的单位硫产酸值相比黄铁矿更高,故精确产酸潜力值反而比全硫预测的产酸值更高。与传统方法计算最大产酸潜力相比,进一步测定矿石中产酸含硫矿物的含量能显著提高产酸潜力预测的准确度。
(3) 测定产酸硫含量的三步连续提取法,适用于以铁硫化物和铜硫化物为主要产酸硫化物的矿石样品。但该方法也存在有待改进的地方和问题:焙烧过程中多种矿物的整体反应尚不完全明确;萃取液分析结果也会受到其他矿物的干扰;对有机硫和未定形态有待更深入的探讨。
