Emergence of highly pathogenic avian influenza A (H5N8) clade 2.3.4.4b viruses in grebes in lnner Mongolia and Ningxia,China,in 2021
2024-01-17QiuziXuXinruYiLiHuaLuoZhenZhangXiangLiRongxiuQinQingAnFengyiQuZhenliangZhaoChengboZhangWeidongWangYuechengLiYajunWangXiangweiZengZhijunHouJingqiangRenYulongWangYanbingLiHongliangChai
Qiuzi Xu ,Xinru Lü ,Yi Li ,Hua Luo ,Zhen Zhang ,Xiang Li ,Rongxiu Qin,Qing An,Fengyi Qu,Zhenliang Zhao,Chengbo Zhang,Weidong Wang,Yuecheng Li,Yajun Wang,Xiangwei Zeng,Zhijun Hou,Jingqiang Ren,Yulong Wang#,Yanbing Li,Hongliang Chai#
1 College of Wildlife and Protected Area,Northeast Forestry University,Harbin 150040,China
2 Harbin Veterinary Research Institute,Chinese Academy of Agricultural Sciences,Harbin 150040,China
3 Wenzhou Key Laboratory for Virology and Immunology,Institute of Virology,Wenzhou University,Wenzhou 325000,China
4 Ordos Forestry and Grassland Administration,Ordos 017000,China
5 Monitoring Center for Terrestrial Wildlife Epidemic Diseases,Yinchuan 750000,China
Highly pathogenic avian influenza (HPAI) subtype H5Nx viruses have spread globally and are a major concern for poultry,wild birds,mammals,and even humans (de Vriesetal.2015;Zengetal.2022).The hemagglutinin (HA)genes of H5 subtype viruses have evolved into multiple clades and some of these clades have been further divided into subclades (Cuietal.2022).Clade 2.3.4.4 H5N8 HPAI viruses (HPAIVs) have caused several waves of disease outbreaks in wild birds and domestic poultry(Wangetal.2022).In the winter of 2013-2014,the HPAI H5N8 virus belonging to clade 2.3.4.4 was first reported as the cause of large outbreaks in poultry in South Korea.The virus subsequently spread to Japan,Russia,Western Europe,and North America (GCH5N8RIV 2016).In 2016,novel HPAI H5N8 viruses of clade 2.3.4.4 were identified in wild birds at Uvs Nuur Lake (Russia) (Leeetal.2017) and Qinghai Lake (China) (Lietal.2017).These viruses reassorted with low pathogenic avian influenza(LPAI) viruses and spread to Europe,Asia,and Africa in the winter of 2016-2017 (Fusaroetal.2017;Kimetal.2017;Yehiaetal.2018).After small outbreaks of H5N8 were reported in the European poultry sector in early 2020 (Smietankaetal.2020),HPAI H5N8 viruses were continuously detected in Iraq,Russia,and Germany in the autumn (Kingetal.2021;Lewisetal.2021;Pyankovaetal.2021).During the winter of 2020-2021,novel H5N8 viruses were introduced into China by wild swans and spread to other species of wild birds and poultry across a wide geographic area (Lietal.2021;Liuetal.2021;Zhangetal.2021;Cuietal.2022).
Wild species ofAnseriformesare often considered to be the important driving force for the cross-regional spread of avian influenza viruses (Websteretal.1992).Long-distance migratory birds play an important role in the inter-regional spread of the HPAI H5N8 virus (GCH5N8RIV 2016).However,the ecological role of short-distance migratory wild birds or resident birds in spreading influenza virus is unclear.Grebes are widely distributed in China.Most overwinter in southern China (Guangdong,Hunan and other places) and breeding sites are generally located in Inner Mongolia and Northeast China (Songetal.2022).Here,we report the detection of HPAI A(H5N8) in dead grebes and environmental samples collected in central China during May-June 2021.We sequenced and analyzed the full-length genomes of 10 H5N8 isolates to track their origin and to evaluate the importance of grebes for spread of the HPAI H5 virus in breeding grounds.
On 14 May,2021,one dead black-necked grebe(Podicepsnigricollis) was found in the Hongjiannaor wetland in Ordos City,Inner Mongolia,China.Tissue samples (trachea,liver,lung,pancreas,kidney,spleen,and rectum) were collected.A total of 143 environmental fecal samples were also collected from the surrounding wetland.In June 2021,dozens of black-necked grebes and great crested grebes (P.cristatus) were found dead in Nanhu wetland in Yinchuan City,Ningxia,China.Organ samples from six dead grebes were collected.All samples were stored at 4°C until they were transported to the laboratory,where they were subsequently stored at-80°C until virus isolation.
Samples were placed in phosphate-buffered saline supplemented with penicillin,streptomycin,and 10%glycerin and oscillated for 1 min.The supernatant from samples was inoculated into 10-day-old specificpathogen-free chicken embryos.After 72 h of incubation at 37°C,allantoic fluid was harvested and HA activity was assayed.RNA was extracted from HA-positive samples using a QIAamp Viral RNA Mini Kit (QIAGEN,Valencia,CA,USA) according to the manufacturer’s instructions and reverse transcribed using the primer 5´-AGCRAAAGCAGG-3´.Studies with highly pathogenic H5N8 avian influenza viruses were conducted in a biosecurity level 3+facility approved by the Ministry of Agriculture and Rural Affairs of China at the Harbin Veterinary Research Institute (HVRI) of the Chinese Academy of Agricultural Sciences (CAAS).
The eight fragments of the H5N8 virus were sequenced using a set of specific primers (Chaietal.2022) and amplified using TaKaRaExTaq(TaKaRa Bio Inc.,Kusatsu,Japan) according to the manufacturer’s instructions.The sequencing data were compiled using the SeqMan Program (DNAStar,Madison,WI,USA).
A BLAST search was performed against the sequencing data in the Global Initiative on Sharing Avian Influenza Data (GISAID) EpiFlu database and GenBank to identify and download the closest relatives of our isolates.Phylogenetic analyses were performed for all eight gene segments.The H5 phylogenetic analyses were conducted with an integrated dataset comprising our isolates and global HPAI H5 viruses in GISAID and GenBank during January 2020 and August 2021.The sequences were alignedviamultiple alignment using fast Fourier transform (MAFFT) (Rozewickietal.2019) implemented in PhyloSuite (Zhangetal.2020).Maximum likelihood phylogenies were inferred using IQ-TREE (Nguyenetal.2015) under the best-fit substitution model for 10,000 ultrafast bootstraps (Minhetal.2013).The best-fit substitution model was selected using the Bayesian information criterion in ModelFinder(Kalyaanamoorthyetal.2017).To better observe the genetic relatedness of the H5N8 isolates,closely related viruses with full genome sequences were assembled and analyzed using the median-joining method implemented in NETWORK5.0 (Bandeltetal.1999).
The avian influenza virus (AIV)-positive host for environmental samples were identified by amplifying cytochromebsequences,as described previously by Chaietal.(2022).In brief,host DNA was extracted using a QIAamp Fast DNA Stool Mini Kit (Qiagen,Hilden,Germany) according to the manufacturer’s instructions.The bird universal primers used were LTyr(5´-TGTAAAAAGGWCTACAGCCTAACGC-3´) and COI907aH2 (5´-GTRGCNGAYGTRAARTATGCTCG-3´)(Tavares and Baker 2008).
For the Inner Mongolia samples,one H5N8 virus from the dead black-necked grebe and three H5N8 viruses from 143 fecal samples were isolated.The species origin of the fecal samples was identified as Eurasian spoonbill(Platalealeucorodia).In addition,six H5N8 viruses were isolated from the Ningxia dead grebes (Appendix A).The HA cleavage site of 10 H5N8 isolates was identified as REKRRKR↓GLF,which is defined as a molecular marker of HPAI viruses.All isolates shared high nucleotide identity across the eight gene segments (99.4-100%),except for the isolate A/Eurasian spoonbill/Inner Mongolia/E42/2021(H5N8) (E42),which only shared 92.9-93.0% identity with nucleoprotein (NP) genes.All 10 viruses harbored the amino acid mutations S133A,T156A,and G182V in the HA protein.These amino acids have been reported to increase the affinity to human-like (α2-6-linked sialic acid)receptors (Yangetal.2007).The viruses also harbored K389R,K482R,and V598T mutations in PB2 protein,N30D and I43M mutations in M1 protein,and P42S and C138F mutations in NS1 protein.The findings suggest an increase in replication and transmission within mammals(Jiaoetal.2008;Fanetal.2009;Huetal.2017;Lietal.2018;Suttieetal.2019).Other key molecular markers of the H5N8 viruses isolated in IM-H5N8 and NX-H5N8 are listed in Table 1.
In the autumn of 2020,clade 2.3.4.4b H5N8 viruses were reportedly introduced into China and two geneticallydistinct lineages (2.3.4.4b1 and 2.3.4.4b2) were confirmed(Lietal.2021;Li Xetal.2022;Li Yetal.2022;Lvetal.2022).The phylogenetic tree showed that our 10 isolates all fall within clade 2.3.4.4b2 (Fig.1-A).Excepting the NP gene of E42,all the isolates clustered together with H5N8 viruses prevalent in China in autumn 2020 at the genome level (Appendix B).The NP gene of E42,which clustered with the LPAI virus prevalent in waterfowl in East Asia during 2019 and 2020,shared the highest nucleotide identity with A/duck/Mongolia/419/2019 (H5N3) (99.4%)and A/duck/Bangladesh/17D1736/2021 (H2N1) (99.3%).These results suggest that HPAI H5N8 viruses have persisted in China since the autumn of 2020 and reassorted with waterfowl-origin LPAI virus during circulation.
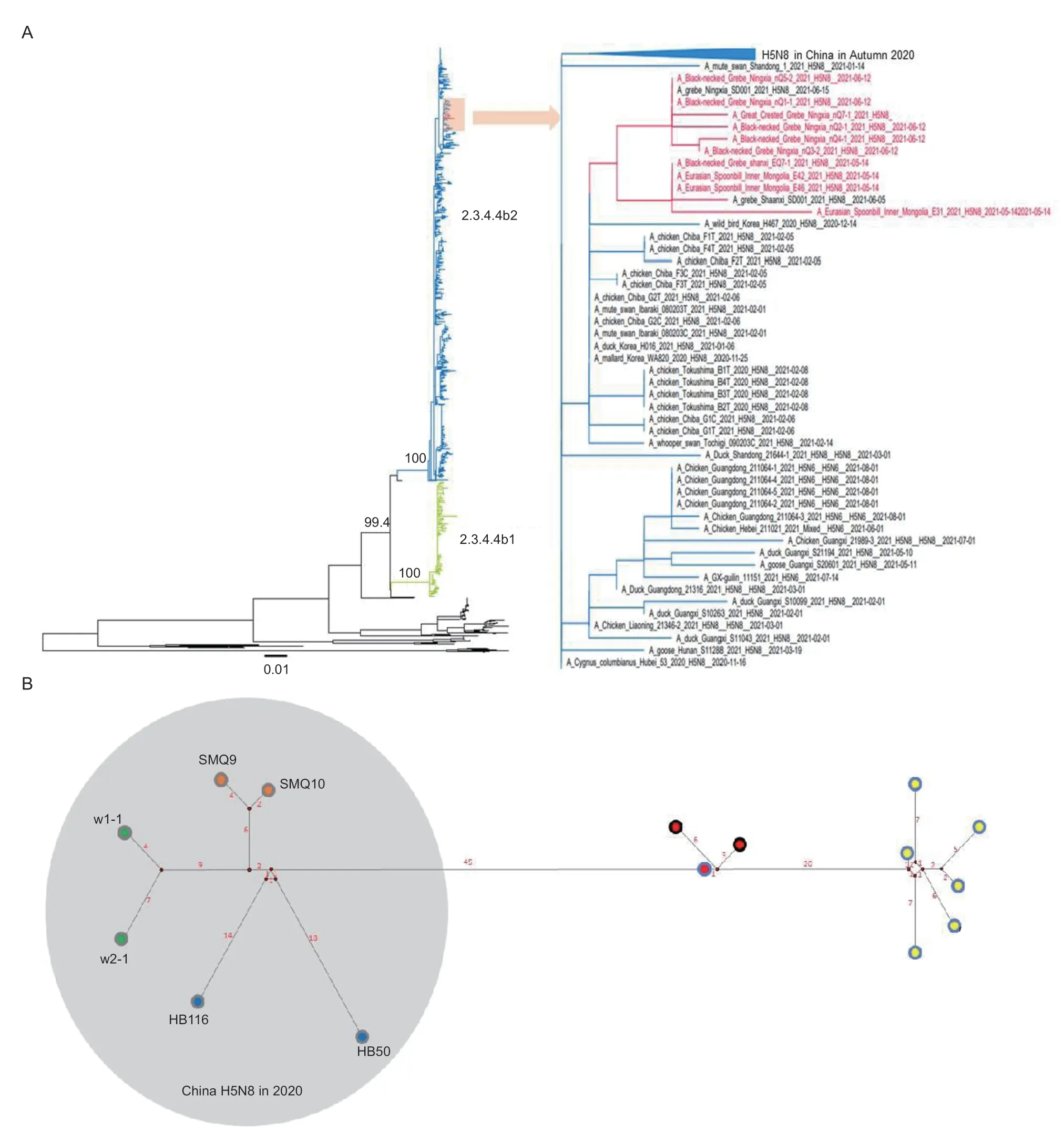
Fig.1 The maximum likelihood (ML) phylogenetic analysis and median median-joining phylogenetic network of H5N8 HPAIVs in Inner Mongolia and Ningxia,China.A,maximum likelihood phylogenetic trees for hemagglutinin genes of avian influenza A (H5N8)viruses.Black lines indicate viruses belonging to clade 2.3.4.4a,c-h.Green and blue branches indicate subclade 2.3.4.4b1 and subclade 2.3.4.4b2,respectively.The subtrees on the right highlight the H5N8 isolates in this study (colored in red) and the H5N8 viruses isolated in East Asia in the autumn of 2020.UFBoot support values of the major branch are indicated.Scale bars indicate nucleotide substitutions per site.B,median-joining phylogenetic network of influenza A (H5N8) viruses identified in China in 2021.The median-joining network was constructed from concatenated H5N8 virus genomes containing all eight segments.This network includes all the most parsimonious trees linking the sequences.Each unique sequence is represented by a circle size relative to its frequency in the dataset.Isolates are colored according to the sample: a green inner circle represents H5N8 reported in Inner Mongolia during October 2020,an orange inner circle represents H5N8 in Henan (wintering ground in the Yellow River basin),a blue inner circle represents H5N8 in Hubei (wintering ground in the Yangtze River basin),a red inner circle represents H5N8 in Inner Mongolia in this study,and a yellow inner circle represents H5N8 in Ningxia in this study.A gray outer circle represents isolates from Anatidae,a black outer circle represents isolates from Eurasian spoonbill,and a blue outer circle represents isolates from grebe.Red numbers indicate the number of nucleotide changes between isolates.Black numbers are abbreviated isolate names.

Table 1 Key molecular markers of the H5N8 viruses isolated in Inner Mongolia and Ningxia,China,in May-June 2021
The H5N8 isolates described here shared a common ancestor with H5N8 strains circulating in China during the autumn of 2020.Thus,we selected six H5N8 viruses reported in autumn 2020 in China and our isolates for further phylogenetic network analyses.The network spectrum showed that after first detection of the H5N8 virus in swans in Inner Mongolia during October 2020,viruses with minor differences were subsequently detected in wintering grounds (Henan and Hubei).However,a series of nucleotide substitutions (45 nucleotides) were observed when we isolated the similar H5N8 virus in Inner Mongolia in May 2021 (Fig.1-B).At that time,the H5N8 virus had first been isolated from dead blacknecked grebes in Inner Mongolia.Similar viruses with minor differences were also detected in surrounding fecal samples from Eurasian spoonbill.Approximately 1 month later,428 km away from the Hongjiannaor wetland,dozens of grebes died of H5N8 virus infection in Ningxia.More than 20 nucleotide shifts were observed (Fig.1-B).
Inner Mongolia and Ningxia are located in the East Asian-Australasian flyway (Tianetal.2015),and are important stopover sites for migratory birds and breeding sites for some migratory birds,including grebes.Approximately 100 grebes died due to infections with clade 2.3.2.1c H5N1 virus in Inner Mongolia in May 2015(Bietal.2016).In May 2016,great crested grebes died due to H5N8 virus infection at Uvs Nuur Lake in Russia(Marchenkoetal.2017).In this study,we observed that among the many wild birds sympatric in the breeding grounds,only grebes suffered serious mortality from HPAI H5N8 viruses.During sampling,ataxia,facial twitching,tilted head,and neck twisting were evident in infected grebes.These neurological symptoms are early clinical signs of AIVs.The results demonstrate that grebes may be susceptible to HPAI H5N8 virus and that grebes infected with HPAI H5N8 virus may show clinical signs that can include death.Considering the sensitivity of grebes to the HPAI H5N8 virus,grebes could be an indicator species for monitoring the invasion of this virus.Enhanced active surveillance of grebes is necessary.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (31970501),the Fundamental Research Funds for the Central Universities,China(2572022CG01),and the National Forestry and Grassland Administration,China.We thank International Science Editing (http://www.internationalscienceediting.com) for editing this manuscript.
Declaration of competing interest
The authors declare that they have no conflict of interest.
Ethical approval
The study has no ethical implications.
Appendicesassociated with this paper are available on https://doi.org/10.1016/j.jia.2023.09.026
杂志排行

Journal of Integrative Agriculture的其它文章
- Can whole steps of grain production be outsourced? Empirical analysis based on the three provinces of Jiangsu,Jilin,and Sichuan in China
- Promoting grain production through high-standard farmland construction: Evidence in China
- The protective effect of cyclodextrin on the color quality and stability of Cabernet Sauvignon red wine
- Quantification of the adulteration concentration of palm kernel oil in virgin coconut oil using near-infrared hyperspectral imaging
- Quantifying the agreement and accuracy characteristics of four satellite-based LULC products for cropland classification in China
- The competition between Bidens pilosa and Setaria viridis alters soil microbial composition and soil ecological function