3D genome organization and its study in livestock breeding
2024-01-17JieChengXiukaiCaoShengxuanWangJiaqiangZhangBinglinYueXiaoyanZhangYongzhenHuangXianyongLanGangRenHongChen
Jie Cheng ,Xiukai Cao ,Shengxuan Wang ,Jiaqiang Zhang ,Binglin Yue ,Xiaoyan Zhang ,Yongzhen Huang,Xianyong Lan,Gang Ren,Hong Chen,
1 Key Laboratory of Animal Genetics,Breeding and Reproduction of Shaanxi Province,College of Animal Science and Technology,Northwest A&F University,Yangling 712100,China
2 Joint International Research Laboratory of Agriculture and Agri-Product Safety of Ministry of Education of China,Yangzhou University,Yangzhou 225009,China
3 College of Animal Science,Xinjiang Agricultural University,Urumqi 830052,China
Abstract Eukaryotic genomes are hierarchically packaged into cell nucleus,affecting gene regulation.The genome is organized into multiscale structural units,including chromosome territories,compartments,topologically associating domains(TADs),and DNA loops.The identification of these hierarchical structures has benefited from the development of experimental approaches,such as 3C-based methods (Hi-C,ChIA-PET,etc.),imaging tools (2D-FISH,3D-FISH,Cryo-FISH,etc.) and ligation-free methods (GAM,SPRITE,etc.).In recent two decades,numerous studies have shown that the 3D organization of genome plays essential roles in multiple cellular processes via various mechanisms,such as regulating enhancer activity and promoter-enhancer interactions.However,there are relatively few studies about the 3D genome in livestock species.Therefore,studies for exploring the function of 3D genomes in livestock are urgently needed to provide a more comprehensive understanding of potential relationships between the genome and production traits.In this review,we summarize the recent advances of 3D genomics and its biological functions in human and mouse studies,drawing inspiration to explore the 3D genomics of livestock species.We then mainly focus on the biological functions of 3D genome organization in muscle development and its implications in animal breeding.
Keywords: 3D genome organization,3D genomic methodology,regulatory mechanisms,muscle development,livestock breeding
1.Instruction
The nucleosome is the fundamental chromatin unit in eukaryotic cells,containing histone proteins and~147 bp of double-stranded DNA.The DNA is wrapped around the histone proteins to form the first chromatin organization level in the mammalian cell nucleus.Higherorder chromatin structure is believed to underlie this phenomenon,packing 2-m-long DNA molecules into~10-μm cell nuclei.The recent development of cutting-edge methodologies and sequencing technologies has shown the three-dimensional (3D) genome structure to be highly dynamic and provide a structural basis for multiple cellular biological processes,such as the cell cycle,cell division,and cell differentiation,by regulating gene expression.
Interestingly,3D genome structures associated with enhancers or promoters have been widely used to explain associations between non-coding genome-wide association study (GWAS) hits and phenotypic variation or complex diseases based on spatial interactions.The Encyclopedia of DNA Elements (ENCODE) project has identified hundreds of thousands of functional DNA elements in the human genome to date (Consortium 2012;Davisetal.2018).In addition,the International Human Epigenome Consortium (IHEC) coordinated the creation of a catalog of high-resolution reference epigenomes for major primary human cell types (Stunnenbergetal.2016).However,it is challenging to systematically and effectively identify their target genes using traditional genomic assays based on linear gene and element positions.The concern is that enhancers can function at various distances by forming chromatin loops to bring them proximal to their target gene.Therefore,the 3D genome structure provides valuable information on interactions between regulatory elements and their target genes.Indeed,forming chromatin folds into specific 3D structures,making distant regulatory elements in the linear genome sufficiently close to their target genes,exerts precise regulatory effects (Albert and Kruglyak 2015).
With progress in 3D genomics,researchers realized that precise,long-distance gene expression regulation is ubiquitous in the mammalian genome (Kragesteenetal.2018).For example,the MYC proto-oncogene bHLH transcription factor (MYC) and Pvt1 oncogene (PVT1)promoters can competitively interact with four enhancers within thePVT1gene region.Mutations in thePVT1promoter enhance the interaction between theMYCpromoter and its enhancer,promoting cancer susceptibility(Choetal.2018).Obesity-related variants in the introns of the FTO alpha-ketoglutarate-dependent dioxygenase(FTO) gene interact distantly with the Iroquois homeobox 3 (IRX3) gene promoter (Smemoetal.2014).
Furthermore,recent studies have also shown that tissue-specific transcription factors (TFs) play important roles in a given tissue’s 3D genome organization.For example,long-range chromatin interactions centered on paired box 3 (Pax3)-bound elements recruit LIM domain binding 1 (Ldb1) for embryonic myogenesis activation(Maglietal.2019).An uncharacterized paired box 7(Pax7)-bound enhancer hub was validated and found to regulate essential myosin heavy chain clustering during skeletal muscle cell differentiation (Zhangetal.2020).Myogenic differentiation 1 (MYOD1) TF reconfigures the 3D chromatin architecture to control gene expression,a process otherwise achieved by the combined activities of multiple TFs (Dall’Agneseetal.2019).
Despite the findings described above,the mechanisms underlying the formation of hierarchical genome structures and the competing structure-function priorities remain incompletely understood.Answering these questions may facilitate genetic explanations for phenotypic variations and complex diseases and their potential applications in animal breeding.
Indeed,livestock husbandry plays a critical role in improving human well-being.In addition,livestock phenotypic variations are associated with non-coding regions identified by GWAS whose mechanistic bases urgently need to be determined.Regulatory elements were recently identified in chickens,cattle,and pigs(Halsteadetal.2020;Caoetal.2021;Kernetal.2021).Moreover,these identified regulatory elements were examined using gene expression atlases,GWAS,and selection signatures for these species (Fangetal.2020;Liuetal.2020),providing insights into the relationship between gene regulation and traits.
In addition to the direct methods to detect regulatory elements mentioned above,some indirect methods also exist.For example,sequencing and comparative analysis of 29 eutherian genomes,including cow and horse,reported nearly a million elements overlapping potential promoter,enhancer,and insulator regions (Lindblad-Tohetal.2011).In addition to traditional comparative genomic analysis methods,machine learning classification methods can identify potential active regulatory regions(promoters,enhancers,and TF binding sites) in livestock species (e.g.,cows and pigs) (Nguyenetal.2018).
Using only comparative genomic analysis to predict regulatory DNA sequences is inadequate.Experiments using methods such as chromatin immunoprecipitation sequencing (ChIP-Seq) are required to confirm them.Candidate bovine enhancer regions identified using publicly available bovine ChIP-Seq enhancer data confirmed enhancers identified based on sequence homologies with human and mouse enhancer databases(Wangetal.2017).In addition,comparative functional annotation was performed for sheep by comparing the genome sequences of 44 breeds and combining these data with experimental ChIP-Seq in sheep tissue to identify active sheep promoters,enhancers,and repressors (Naval-Sanchezetal.2018).
The 3D structure of genome has been established as a crucial regulator of gene expression through various mechanisms.Various techniques,summarized as imaging,3C-based,and ligation-free methods,have been developed to study the 3D genome and its function,with potential for further advancements.The 3D genome study has been expanded beyond model organisms to livestock,and proven to be a valuable tool for interpreting key functional mutations in the post-GWAS era.Here,we review the hierarchical structures of the 3D genome,their mechanisms of formation in eukaryotic cells (the toolbox for 3D structure identification),and their mechanisms of action in gene regulation.We provide a comprehensive picture of the 3D genome and highlight its potential implications for muscle development.Finally,we examine the current state of 3D genomics in livestock and summarize current knowledge to improve our understanding of trait-associated gene regulation in the context of animal breeding.The 3D genome can help identify functional regulatory elements and advantageous mutations to explain the molecular mechanisms underlying phenotypic variations,making it a topic of interest in the postgenomics era.Therefore,systematic analyses of 3D genome structures will provide a basis for the precise breeding and genetic improvement of farm animals.
2.Hierarchical 3D genome organization
The genome is hierarchically organized into multiscale 3D structural units,including chromosome territories,transcriptionally active/inactive (A/B) compartments,topologically associating domains (TADs),and DNA loops.
2.1.Chromosome territories
The genome is not randomly distributed across the nuclear volume.Microscopic observation and chromatin conformation capture techniques have shown that each chromosome has significant self-interactions and occupies a different area,known as a chromosome territory (Fig.1) (Cremer and Cremer 2010;Naganoetal.2017).Interestingly,gene-enriched regions were thought to be located at chromosome territory boundaries based on a small number of genes,whereas comprehensive analysis of nuclear transcription after labeling of nascent transcripts with Br-UTP showed incorporation throughout the chromosome territory (CT) (Branco and Pombo 2006).Also,inter-chromosomal interactions (i.e.,transinteractions) are intensively observed at chromosome territory (Stevensetal.2017;Shahetal.2018).

Fig.1 Hierarchical 3D genome organization.Intra-chromosome interactions are generally more common than inter-chromosome interactions and occupy distinct regions within the nuclear volumes,known as chromosome territories.B compartments near the nucleolus and nuclear lamina have low transcriptional activity.In contrast,those between them and near the nuclear speckle have high transcriptional activity and are called A compartments.Currently,there are two different 3D genome models.Generally,one is that compartment is larger than topologically associating domain (TAD),and the other is that compartment domain within TAD.However,the former may be more stable than the latter,given a higher frequency of loops in the interaction heatmap.Detecting interactions between promoters and enhancers often requires high sequencing depth and resolution.The interactions between promoters and enhancers can be mediated by CCCTC binding factor (CTCF) or other factors such as YY1.The nuclear lamina is a structure near the inner nuclear membrane and the peripheral chromatin and it is composed of lamins,which are also present in the nuclear interior,and lamina-associated proteins.At the nuclear periphery,associations of chromatin with the nuclear lamina through lamina-associated domains (LADs) aid functional organization of the genome.Nuclear speckles,also known as interchromatin granule clusters,are nuclear domains enriched in pre-mRNA splicing factors,located in the interchromatin regions of the nucleoplasm of mammalian cells.Nuclear pore complexes (NPCs) are the sole gateways that facilitate this macromolecular exchange across the nuclear envelope with the help of soluble transport receptors.The nuclear envelope (NE) is a highly regulated membrane barrier that separates the nucleus from the cytoplasm in eukaryotic cells and it contains a large number of different proteins that have been implicated in chromatin organization and gene regulation.
2.2.Chromosome A/B compartments
The chromosome A/B compartments were first discovered in the human genome using high-throughput chromosome conformation capture (Hi-C) at a 1-Mb resolution (Lieberman-Aidenetal.2009).Integrating compartments with known genetic and epigenetic features showed that they have transcriptionally active (A) and inactive (B) forms that alternate across the genome(Fig.1).Compartment A regions have higher gene densities,active epigenetic marks,and high transcription activities.In contrast,compartment B regions have higher transposable element densities and repressive epigenetic marks (Lieberman-Aidenetal.2009;Padeken and Heun 2014;Stevensetal.2017).Genomic regions with similar chromosomal characteristics significantly interact,with A-A and B-B interactions more frequent than A-B interactions (Lieberman-Aidenetal.2009).These interaction profiles show a lattice pattern in the Hi-C contact matrix (Lieberman-Aidenetal.2009;Wangetal.2016).
Recent studies have shown that chromosome A/B compartments have a biased nuclear distribution.A compartments are often close to nuclear speckles,protrusive nucleosomes containing enhancer RNAs(eRNA) and active transcriptional factors.In contrast,B compartments are often near lamina-association domains and nucleoli (Zheng and Xie 2019;Goel and Hansen 2020;Wangetal.2021) (Fig.1).Interestingly,depletion of the structural protein serine/arginine repetitive matrix 2 (Srrm2) in nuclear speckles decreased A compartment strength but increased B compartment strength in Hi-C(Huetal.2019),suggesting that their formation may be antagonistic.
The nuclear lamina is a mesh structure comprising lamins and lamin-associated proteins found inside the nuclear membrane (Hildebrand and Dekker 2020) and around the nucleolus,where inactive inter-chromosomal hubs,centromeres,and ribosomal DNA regions are found (van Koningsbruggenetal.2010;Némethetal.2010;Pontvianneetal.2016).B compartments are bound by the polycomb repressive complex,contributing to developmental gene suppression,and contain heterochromatin protein 1 (HP1),driving heterochromatin formation (Hildebrand and Dekker 2020).Lamins A/C and polycomb group (PcG) play a crucial role in muscle development.For example,muscle stem cells (MuSCs)lacking lamin A/C redistribute PcG-dependent histone marks,leading to the transcriptional upregulation of crucial PcG-target genes,including non-muscle-related genes and cell cycle inhibitors (e.g.,cyclin-dependent kinase inhibitor 2A [CDKN2A/p16Ink4a]) (Bianchietal.2020).
2.3.TADs
TAD is a self-interacting domain formedviaa loopextrusion mechanism.It was originally defined as Mbscale genomic blocks in low-resolution (40 kb) mammalian Hi-C matrices.Higher interaction frequencies were observed inside than outside TADs (Fig.1) (Dixonetal.2012),potentially reflecting that TADs are separated from the adjacent region by an obvious boundary,forming an independent regulatory unit whose primary function is to limit the interaction distance between regulatory elements(Symmonsetal.2014).
TAD boundaries sometimes have significant interactions shown as distant vertices in Hi-C heatmaps due to the transient nature of loop extrusions in millions of cells.In addition,TAD boundaries are generally more highly conserved across species than loops (Eres and Gilad 2020).Moreover,TAD boundaries typically have histone modifications associated with gene activation,such as H3K4me3 and H3K36me3 (Raoetal.2014).Besides,many chromatin structural proteins can bind to the TAD boundary and play important roles in TAD structure and stability,including CCCTC binding factor(CTCF) and cohesin (Li Metal.2020).
The development of single-cell omics,including singlecell Hi-C,indicated that TADs were not identical among single cells (Tanetal.2018),with a small subset of TAD boundaries cell-type-specific (Dixonetal.2015).For example,single-cell Hi-C in mice showed that TADs were present in single cells but differed between cells (Naganoetal.2013).Single cells still had TAD-like structures without CTCF and cohesin bound to TAD boundaries.It is unclear whether there is a fundamental difference between cohesin-dependent and -independent TADs.
What is the relationship between compartments,compartmental domains,and TADs? Compartments were initially identified in 1-Mb binned Hi-C heatmaps,indicating that multiple Mb-scale TADs are nested within a single contiguous A or B compartment segment.However,increasing Hi-C sequencing depth enabled Lieberman-Aidenetal.(2009) to identify interacting domains smaller than TADs.Rowleyetal.(2018) subsequently defined these structures as compartmental domains that directly result from establishing A/B compartments and are defined by their internal chromatin state.Based on this finding,Rowleyetal.(2017) proposed that TADs are created through CTCF-looping of compartmental domains.Therefore,TADs may contain different compartmental domains,as shown in Fig.1.
In conclusion,there are currently two different 3D genome models (Fig.1;MODEL1 and MODEL2).Generally,one is that compartment is larger than TAD,and the other is that compartment domain within TAD.However,the former may be more stable than the latter,given the higher frequency of loops in the interaction heatmap (Razin and Ulianov 2017;Rowley and Corces 2018;Beagan and Phillips-Cremins 2020).In addition,TADs and compartments share similar chromatin features inAnophelesmosquitos (Lukyanchikovaetal.2022).
2.4.Chromatin loop and extrusion models
Chromatin loops form by physical interactions between two genomic loci and present as ‘corner dots’,a punctated group of adjacent pixels with significantly enhanced interaction frequencies than their surrounding pairwise loci in a heatmap.Chromatin loops can be broadly categorized into structural and functional loops.Structural loops usually function in TAD formation and are sometimes much larger than functional loops.In contrast,functional loops are typically hundreds of kb and commonly mediate contacts between regulatory elements (Spitz 2016;Wangetal.2021).These loopmediated interactions are commonly restricted to within a TAD,insulating interactions with neighboring domains,and TAD disruptions pathogenically rewire gene-enhancer interactions.Deep sequencing could produce highresolution Hi-C data,essential for identifying interaction loops,which is the primary focus of 3D genome research since DNA loops identify the target genes of regulatory elements (Schoenfelder and Fraser 2019).
Chromatin loops show high-frequency interaction peaks in the Hi-C interaction matrix (Raoetal.2014)mainly due to loop extrusion,which facilitates enhancerpromoter interactions.In the loop extrusion model,the cohesin complex can form a circular structure,recruiting the NIPBL cohesin loading factor and MAU2 sister chromatid cohesin factor to facilitate its loading onto chromatin.Extrusion is blocked until cohesin reaches the chromosomal boundary formed by forward and reverse CTCF (Ganjietal.2018).Finally,the cohesin complex releases CTCF from chromatin through the WAPL and PDS5 cohesin release factors (Haarhuisetal.2017).In addition,gene transcription facilitates cohesin translocation,promoting loop formation (Heinzetal.2018).Therefore,several loop structures form inside the TAD,promoting interactions within it.
CTCF and cohesin can promote TAD and loop formation through the loop extrusion model (Fudenbergetal.2016),stabilize enhancer-promoter interactions,and maintain robust gene expression (Renetal.2017).It should be noted that CTCF binding sites at the two loop anchors are usually in a convergent linear orientation (Raoetal.2014),and changing the CTCF motif direction would impair loop and TAD formation (Guoetal.2015).In addition,the deletion of the loop extrusion factor,cohesin complex subunits,or its loading factor NIPBL resulted in extensive TAD and loop loss or reduction (Schwarzeretal.2017).However,a recent circular chromosome conformation capture (4C) study found that knocking out the CTCFassociated TAD boundaries did not affect the local chromatin interaction pattern within TADs (Simaetal.2019).Other factors besides CTCF and cohesin may contribute to these local chromatin interactions.The YY1 transcription factor has been reported to promote interaction loop formation.However,this type of loop may be less stable than the CTCF loop,which usually presents as corner dots in heatmaps (Fig.1) (Spitz 2016;Wangetal.2021).
3.Methods for 3D genome architecture mapping
Our knowledge about orderly genomic chromatin compaction depends on technical innovations.Until recently,two main technologies have been widely used in this field: imaging techniques,particularly DNAfluorescenceinsituhybridization (FISH) and Hi-C.Both techniques can map local interactions between regulatory elements.Some variations of these techniques can map genome-wide interactions while simultaneously revealing the genome’s 3D hierarchy.
3.1.Imaging methods
FISH is the original method for examining chromatin spatial structure and genome segment interactions.It uses fluorescently-labeled DNA oligonucleotides as probes that hybridize to complementary genome regions.DNA-FISH is commonly used to measure the physical distances of 2-52 target genomic regions (Kempfer and Pombo 2019).A distance threshold usually defines chromatin contact,and fluorescence signals are co-localized over a spatial distance of 50 nm to 1 μm (Barbierietal.2017;Barutcuetal.2018;Maassetal.2018).Unfortunately,DNA-FISH cannot identify all pairwise interactions among genomewide loci.
Unlike 2D-FISH,3D-FISH can detect 3D spatial structures.Probes typically cover genome sequences ranging from 30 kb to an entire chromosome.Therefore,large genomic regions,such as TADs (Noraetal.2012;Beagrieetal.2017) or entire chromosomes (Branco and Pombo 2006),can be accurately detected by standard 3D-FISH.However,FISH’s signal-to-noise ratio increases with target length due to greater localized fluorescence and specificity.Interactions between chromosome regions<100 kb are challenging to detect with 3D-FISH,such as enhanced-promoter interactions.For example,DNA-FISH was used to detect MYC and bromodomain containing 4(BRD4) colocalization after enhancer perturbation in K562 cells (Linetal.2022).
Cryo-FISH was developed to measure 5-kb regions and provide high-resolution and precise chromatin contact imaging (Beliveauetal.2015).Standard FISH probes are hybridized with cell-fixed thin (~100-200 nm) cryosections visualized with fluorescence or electron microscopy (Branco and Pombo 2006;Barbierietal.2017;Beagrieetal.2017).In 2D-and 3D-FISH,while nuclear structures and chromatin’s nuclear distribution generally remain intact,the fixation process alters the nucleus’s shape and size,chromatin domain structure,and especially the association of some loci with their chromosome territories (Heppergeretal.2007).In addition,denaturation can disrupt the nuclear membrane,leading to DNA loss (Raapetal.1986).Cryo-FISH uses electron microscopy-grade fixation to preserve nuclear and cytoplasmic ultrastructures and decrease adverse heat denaturation effects.
While FISH-based techniques have been widely used to investigate chromatin architectures,a highly dynamic process during chromosome folding that varies greatly during the cell cycle (Stevensetal.2017;Gibcusetal.2018),their cell fixation requirement prevents their use to dynamically study chromatin activities.Imaging methods have been developed based on genome editing to study chromatin dynamics and allow specific genomic loci to be targeted in living cells.The clustered regularly interspaced short palindromic repeats (CRISPR) system using endonuclease-deficient dead Cas9 (dCas9) fused to a fluorescent protein can locate gene loci in living cells.For example,a genomic locus of interest was visualized through dCas9 fused to an enhanced green fluorescent protein (EGFP) and guided by a sequence-specific small guide RNA (gRNA) to study native chromosome conformation and dynamics in living human cells (Chenetal.2013).In addition,up to six chromosomal loci can be simultaneously imaged in a single cell with the CRISPRainbow system (Maetal.2016).However,because fluorescent signals are relatively weak with these methods,a dCas9-SunTag system with a bright fluorescent protein was developed for live cell imaging (Yeetal.2017).For example,the epigenetic perturbationinduced genome organization (EpiGo)-Krüppel associated box (KRAB) method showed that H3K9me3’s role in genome organization could be partially separated from its gene repression function (Fengetal.2020).
3.2.Chromosome conformation capture (3C)-based methods
The rapid development of high-throughput sequencing technologies enabled Dekkeretal.(2002) to propose a new 3C technology in 2002 that projects 3D interactions on a 2D heatmap through bioinformatics (Dekkeretal.2002).The main 3C analysis steps are as follows:(i) isolate cells,(ii) fix DNA-protein complexes with formaldehyde,(iii) cut the genomic DNA into fragments of specific sizes with enzyme digestion or sonication,(iv) use DNA ligase to form proximal connections,(v)extract DNA,and (vi) perform polymerase chain reaction(PCR) detection.When two sites may remotely interact,divergent primers are designed to amplify across the ligated site.
There exists a large amount of unknown genomic interaction information in 3C library.To explore this information more thoroughly,researchers have proposed several high-throughput techniques based on the 3C technology,such as circular conformation capture (4C),chromosome conformation capture carbon copy (5C),and Hi-C (Dostieetal.2006;Zhaoetal.2006).The Hi-C method was introduced by Lieberman-Aidenetal.(2009)in 2009.It is an improved 3C version that can detect all interactions between two loci in the whole genome(Table 1).Unlike the 3C library,it uses avidin to label enzyme-digested ends,then ligates them using a ligase.Genomic DNA is then extracted,purified,and fragmented into specific sizes by sonication.DNA fragments containing avidin are then enriched and high-throughput sequenced.3D structures can be inferred from the Hi-C interaction matrix.The matrix’s resolution depends on the size of genome fragments (bins) used in the analysis,the endonuclease’s characteristics (4-or 6-base digestion),and sequencing depth.Several software and methods,including HICUP (Wingettetal.2015),HiC-Pro (Servantetal.2015),HiCdat (Schmidetal.2015),HIPPIE (Hwangetal.2015),GOTHiC (Mifsudetal.2017),and Fit-Hi-C2(Kauletal.2020) have been utilized to uncover 3D genome structures,such as compartments,TADs,and loops.
While 3C-based conformation capture and sequencing can confirm the spatial interaction of two remote sites,it cannot study pairwise interactions mediated by specific proteins,such as TFs,chromatin histone modifiers,and architectural proteins.Integrating ChIP with the 3C-based method can perfectly solve this issue.More recently,ChIP-loop,4C-ChIP,chromatin interaction analysis (ChIA)by paired-end tag sequencing (ChIA-PET) and proximity ligation-assisted ChIP-sequencing (PLAC-seq,also known as Hi-ChIP;Table 1) have been developed.ChIP-loop uses specific antibodies to enrich DNA-protein crosslinked complexes and determine whether there is a proteinmediated interaction between the target sites using PCR (Kim and Dekker 2018).While ChIA-PET is similar to ChIP-loop,it can detect many-to-many interactions mediated by specific proteins,unlike ChIP-loop,which can only detect one-to-one interactions (Lietal.2010).In addition,Hi-ChIP (Mumbachetal.2016) and PLAC-seq(Fangetal.2016) have an advantage over ChIA-PET due to their optimized protocol in which ligation occurs within intact nuclei to avoid the bias introduced by enriching for genomic regions bound by the protein of interest (Daviesetal.2017).

Table 1 Comparison of 3C-based methods used to detect chromatin interaction
3.3.Ligation-free methods
The above methods cannot simultaneously detect multiple DNA interactions within the same nucleus(O’Sullivanetal.2013).Therefore,three ligation-free approaches,genome architecture mapping (GAM),splitpool recognition of interactions by tag extension (SPRITE),and chromatin-interaction analysisviadroplet-based and barcode-linked sequencing (ChIA-Drop),were developed.GAM measures co-segregation frequencies among genomic regions by slicing the nucleus into thin nuclear sections and sequencing the DNA contents of numerous randomly collected slices to map genome-wide chromatin contacts (Beagrieetal.2017;Winick-Ngetal.2021).SPRITE detects entire chromatin interactions among multiple genomic regions by tagging single crosslinked chromatin complexes with unique identifier combinations before sequencing (Quinodozetal.2018).ChIA-Drop integrates the latest microfluidics technology with ChIA and droplet-based and barcode-linked high-throughput sequencing (Zhengetal.2019).These methods are based on the orthogonal ligation method and have begun to provide new insights into 3D genome topology.
Various methods with different strengths and limitations are used to study 3D genome architecture.Imaging approaches (e.g.,2D-,3D-,cryo-FISH) and 3C-based methods (e.g.,3C,4C,5C,and Hi-C) have shown the existence of 3D genome architecture.The recent development of ligation-free approaches (e.g.,GAM and SPRITE) are now uncovering new aspects of the 3D genome and confirming the complex,highly organized nucleus.
4.3D structure regulatory mechanisms on gene expression
The 3D genomic chromatin structures are closely related to dynamic changes in gene expression.For example,chromatin conformation is closely related to dynamic gene expression changes during TF-driven B lymphocyte reprogramming into pluripotent stem cells (Stadhoudersetal.2018).Therefore,how do different genome conformations affect gene expression?
4.1.Chromatin compartments regulate gene expression
A compartments are generally enriched for transcriptionally active genes and have higher gene densities,chromatin openness,and active histone modifications.In contrast,B compartments are generally enriched for transcriptionally repressed genes corresponding to heterochromatin regions (Dongetal.2018;Hofmann 2020;Tian Hetal.2020).Differential genome compartmentalization affected transcriptional heterogeneity among glioblastoma stem cell cultures (Johnstonetal.2019).However,how chromatin compartments regulate gene expression remains unclear since robust evidence based on editing genomic conformations at this level is difficult to achieve.
4.2.TADs regulate gene expression
Structural variants (SVs) located in inter-TAD regions can disrupt TADs or TAD boundaries,resulting in TAD fusion,neo-TADs,TAD shuffling,and the pathogenic rewiring of gene-enhancer interactions.For example,an SV spanning the CTCF-associated boundary elements at the Wnt family member 6 (WNT6),Indian hedgehog signaling molecule (IHH),ephrin receptor A4 (EPHA4),andPAX3locus regulatingEPHA4expression rewires theWNT6,IHH,andPAX3promoter,resulting inEPHA4silencing,abnormalWNT6,IHH,andPAX3expression,and congenital limb malformation (Lupiáñezetal.2015).In addition,an inter-TAD duplication creates a neo-TAD and places potassium inwardly-rectifying channel subfamily J member 2 (Kcnj2) under the control of a sex-determining region Y-box 9 (Sox9) regulatory region,resulting in ectopic contacts,consecutiveKcnj2misexpression,and limb malformation (Frankeetal.2016).Moreover,inversions or translocations due to TAD shuffling can lead to the fusion of two regulatory domains and enhancer adoption.For example,balanced translocations can separate the myocyte enhancer factor 2C (MEF2C) gene from its regulatory enhancer elements,decreasing its expression (Redinetal.2017).Therefore,SV effects on TADs and gene expression regulation must be considered when explaining relationships between TAD and traits.
Finally,the deletion of TAD boundary or loop anchors,such as CTCF,YY1,and the cohesin complex,usually results in irregular gene expression.For example,CTCF knockout causes TAD disappearance,altering the expressions of~2,000 genes (Kuboetal.2021).Similarly,cohesin knockout profoundly impacts the expression of genes regulated by super-enhancers (Stadhoudersetal.2019).Moreover,in muscle development,transcription factor Myod1 can act as an anchor protein and facilitate the formation of myogenesis-specific chromatin loops in muscle cells (Wangetal.2022).In conclusion,CTCF,the cohesin complex,and muscle-specific Myod1 anchors regulate gene expressionviadirecting chromatin interactions.
4.3.Loops regulate gene expression
Manycis-regulatory DNA elements,such as promoters,enhancers,silencers,and insulators,occupy a significant fraction of the mammalian genome,potentially comparable to protein-coding gene sequences.We first focus on enhancer-promoter contacts in gene activation.One of the most prominent examples of long-range enhancers influencing the expression of key genes is a chromosomal loop that brings the bud-specific enhancer,zone of polarizing activity regulatory sequence(ZRS),into proximity with the sonic hedgehog (shh)promoter,activatingshhexpression (Amanoetal.2009).Remarkably,ZRS knockout mice were born with severely truncated limbs (Sagaietal.2005).It is important to note that extensive enhancer-promoter interactions can occur on very short time scales.For example,the dynamic enhancer-promoter interaction changes at the mouse cryptochrome 1 (Cry1) locus control circadian rhythm gene expression (Kim Y Hetal.2018).
Second,promoter-promoter and enhancer-enhancer interactions are also thought to play important roles in gene transcription.For example,extremely long-range promoter-promoter interactions dynamically reorganized between two pluripotency states (Joshietal.2015),and enhancer-enhancer interactions targeting many marker genes contribute to cell identity organization during heart development (Chenetal.2019).
However,many genes,especially those encoding developmental regulators,interact with one or multiple enhancers (Javierreetal.2016),and gene expression levels are significantly positively correlated with enhancer numbers (Javierreetal.2016),suggesting an additive enhancer effect on target gene transcription levels.This property can be interpreted as a hub where a promoter interacts simultaneously with multiple enhancers,increasing the total time for the promoter to interact with one of a range of available enhancers to facilitate gene transcription (Schoenfelder and Fraser 2019).Moreover,one enhancer can regulate multigene promoters without competing.For example,an enhancer upregulates myosin heavy polypeptide 1 (Myh1),3 (Myh3),and 8 (Myh8) gene expression through simultaneous interactions with multi-promoters of theMyhgene family in differentiating iPax7 cells(Zhangetal.2020).
Advanced technologies,such as gene editing,can manipulate the interaction between two genomic loci to disturb the intrinsic promoter-enhancer loop and induce neo-interactions to regulate target gene expression.The dCas9 fusion proteins target two genomic loci to force and engineer chromatin looping,reversibly inducing gene activation by close spatial proximity using small molecule(Morganetal.2017) or light-induced (Kimetal.2019)dimerization.
4.4.eRNA helps stabilize the structure of the 3D genome
Enhancers bound by RNA polymerase II (Pol II) can generate bi-directional non-coding RNAs,called eRNAs (Kimetal.2010;De Santaetal.2010;Lietal.2013;Mousavietal.2013;Lietal.2016).eRNAs are closely related to enhancer function and actincisto stimulate transcription near eRNA loci (Kimetal.2010;Mousavietal.2013;Hsiehetal.2014).Mechanistically,eRNAs can interact with mediator or cohesin complexes to establish or stabilize chromatin looping (Laietal.2013;Lietal.2013).In addition,eRNAs can combine with YY1 to increase its local concentration at loop anchors (Sigovaetal.2015).Furthermore,eRNAs can interact with transcriptional co-activator CREB binding protein (CBP),stimulating core histone acetyltransferase activity and promoting gene expression in a sequenceindependent manner (Boseetal.2017).Studies have shown that eRNAs acting as a decoy for negative elongation factor (NELF) are involved in the productive elongation stage of the transcription process by promoting the release of paused Pol II (Schaukowitchetal.2014).In addition,eRNAs can control transcription elongation by interacting with components of positive transcription elongation factor b(P-TEFb) (Zhaoetal.2016).
In the myogenesis process,Myod1 is a key TF in muscle development,and its distal enhancer can produce eRNAs to enhance the interaction with the cohesin complex and activate myogenin (MYOG) expression (Tsaietal.2018).In addition,lysine demethylase 1A (KDM1A/LSD1) can activate the transcription of eRNAs required for the timely Myod1 expression (Sciontietal.2017) which is crucial for activating eRNA production with positive feedback (Zhaoetal.2019).While our knowledge about eRNAs is growing,efforts are still needed to find other protein binding patterns and eRNA mechanisms of action in regulating gene expression.
In conclusion,a large amount of evidence indicates that forming 3D genome structures can bring enhancers and their target gene promoters into proximity,regulating gene expression (Furlong and Levine 2018).The chromatin loop and TAD formation can effectively limit the promoterenhancer interaction range (Dengetal.2012;Chenetal.2018),which depends on the structural proteins enriched at the anchors of strong chromatin interactions.Finally,eRNAs help stabilize the 3D genome structure to control expression.
5.Perspectives on 3D genomes in livestock breeding
It is estimated that the demand for animal-sourced food will increase by 70% by 2050,requiring the implementation of advanced technologies to improve meat production (Heetal.2018).MuSCs undergoing different fates depend on defined transcriptional hierarchies that ensure the appropriate spatiotemporal regulation of distinct gene sets.Indeed,satellite cell activation is caused by the induction of myogenic regulatory factors such asMYOD1and myogenic factor 5 (MYF5).In contrast,differentiation involves the downregulation ofPAX7,MYOG,and myosin heavy chain(MYHC) expression,followed by the fusion of differentiating myoblasts into myofibers.Pax7 binding to super-enhancers induces dramatic,localized chromatin remodeling characterized by histone mark acquisition,chromatin accessibility induction,and regulatory loop assembly in muscle precursors and lineage-committed myoblasts (Liljaetal.2017;Zhangetal.2020).The MYOD1-directed reconfiguration of chromatin interactions temporally preceded the effect on gene expression and was mediated by direct MYOD1-DNA binding,a process otherwise achieved by the combined activities of multiple TFs (Dall’Agneseetal.2019).
Insulin-like growth factor 2 (IGF2) is a well-known insulin-like growth factor family member that plays an important role in fetal myoblast growth and development,making it one of the most important candidate genes for various economic traits in domestic animals.The DNA methylation-sensitive binding of CTCF at theIgf2-H19locus in mice acts as an insulator to control its epigenetic imprinting (Pombo and Dillon 2015).In addition,CTCFdependent loops are thought to be able to segment chromatin into parent-origin-specific domains at theIgf2-H19locus.Therefore,it is reasonable to hypothesize that disrupting the loop at theIgf2-H19locus will dysregulate key genes in theIgf2-H19locus and cause phenotypic variation in domestic animals.The regulatory elements’ 3D structure is mediated by this interaction loop,including promoter-enhancer,promoter-promoter,and enhancer-enhancer interactions.Consequently,this loop plays a precise role in regulating gene expression.
Besides the identification of loop-mediated regulatory elements,genome haplotype construction and assembly are the leading applications of 3D genomics.Renetal.(2013) have completed a highly accurate (98%) haplotype in human cells.In addition,the HapCUT2 software was explicitly developed for haplotype construction (Edgeetal.2017).Currently,assisted genome assembly has been completed for the goat,pig and other species using nextgeneration sequencing integrated with Hi-C technology to improve genome assembly accuracy (Table 2).
There have been numerous reports about the 3D genomes of various domestic animals.First,3D genomes have been compared between different species.A comprehensive analysis of the 3D genomes of liver and T cells from Holstein cows,Alpine goats,White Leghorn chickens,and Large White pigs has been conducted(Foissacetal.2019).Additionally,a study compared the 3D genomes of the fibroblast cells of fish,chickens,and ten other mammalian species using high-resolution Hi-C data.This study uncovered the genome organization features and the regulation of functional genes across species (Li Detal.2022).Second,studies have been performed specifically on the 3D genome of chickens.The architecture of the 3D genome of chicken liver cells was characterized using the Hi-C technique,comparing Lindian chicken (LDC) liver cells with Wenchang chicken(WCC) liver cells (Shaoetal.2022).Additionally,the first genome-wide analysis of chromatin interactions in chicken embryonic fibroblasts and adult erythrocytes was conducted (Fishmanetal.2019).Third,there is a significant body of research on the 3D genome of pigs.Using theinsituHi-C method,the functional impact of 3D chromatin on the transcriptional regulation of pluripotency marker genes was demonstrated in pig epiblast-derived pluripotent stem cells (Zhietal.2021).Additionally,3D chromatin profiles were generated for various pig cells,including somatic cells and pre-implantation embryos frominvitrofertilization,parthenogenesis,and androgenesis(Li Detal.2020).The 3D genome organization of two adipose tissues was investigated in wild boars and Bama pigs (Zhang Jetal.2022).A recent study revealed the major variations in the 3D genome of myocytes between 90 and 110 days of gestation in pigs (Marti-Marimonetal.2021).A comprehensive interaction map of nuclear dynamics was generatedviachromatin-chromatin(H3K27ac BL-HiChIP) and RNA-chromatin interactions(GRID-seq),revealing genomic variants that contribute to skeletal muscle traits in pigs (Li Metal.2022).The ChIA-PET method was used to study the changes in chromatin 3D structure in the skeletal muscles of lean Yorkshire pigs and fat Meishan pigs (Liu Retal.2022).Furthermore,using various methods including RNA sequencing,ChIP-Seq,ATAC-seq,and Hi-C,Zhao Yetal.(2021) characterized the function ofcis-regulatory elements in pig tissues across 12 diverse tissues.Finally,the 3D genome of cattle has been investigated.Our group has investigated the 3D genome structures in the myocytes of Qinchuan cattle by Hi-C (Fig.2).Our results provide evidence of the relationship between the 3D genome and muscle development.For instance,we have observed that the elevation of tropomodulin 1 (TMOD1)expression during muscle maturation is associated with heightened chromatin accessibility,increased H3K27ac and H3K4me1 levels at enhancers,and the formation of loops (Fig.3).These findings provide a new foundation for understanding the molecular mechanisms underlying muscle development in livestock.
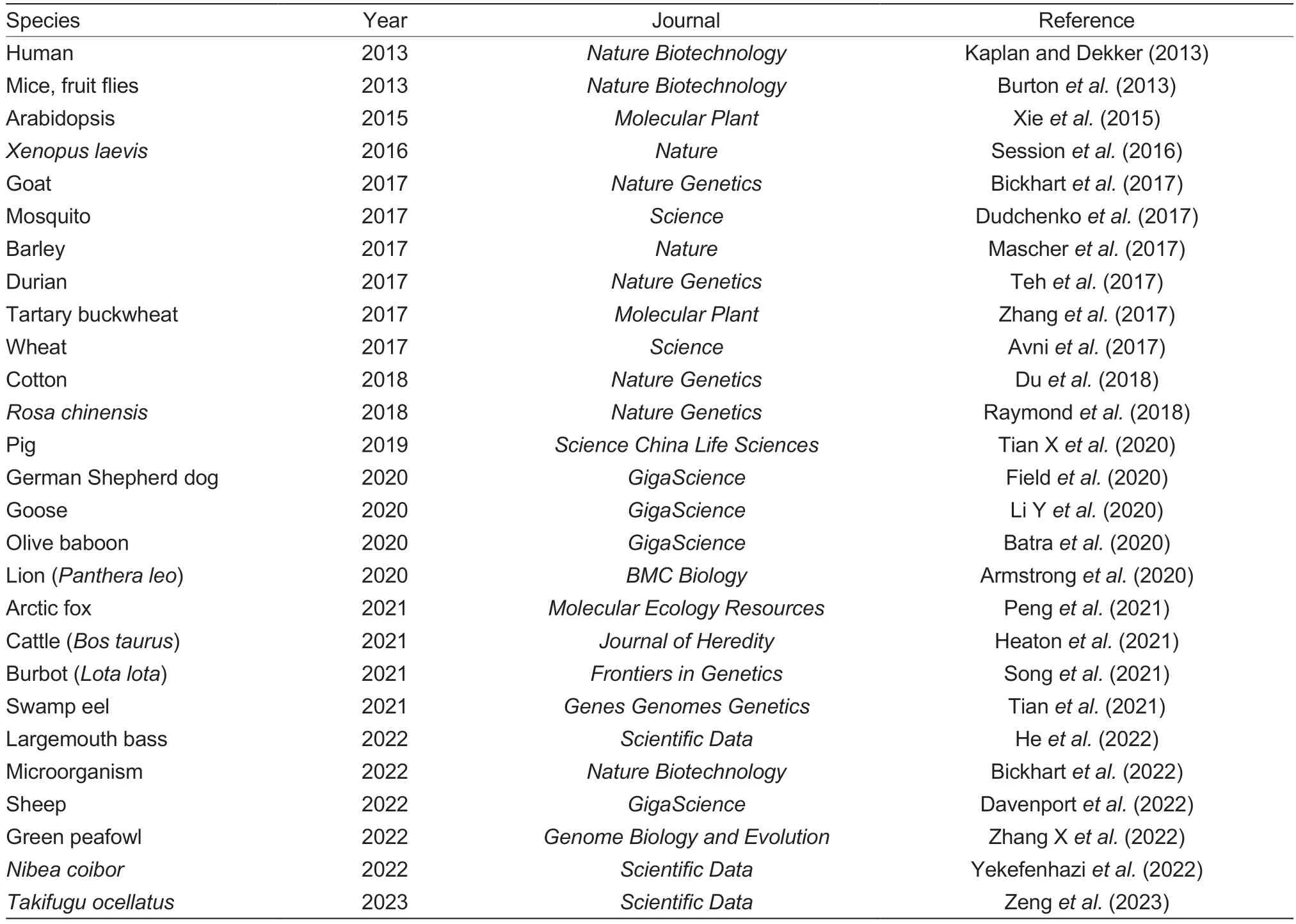
Table 2 Proceedings of genome assembly with Hi-C for various species
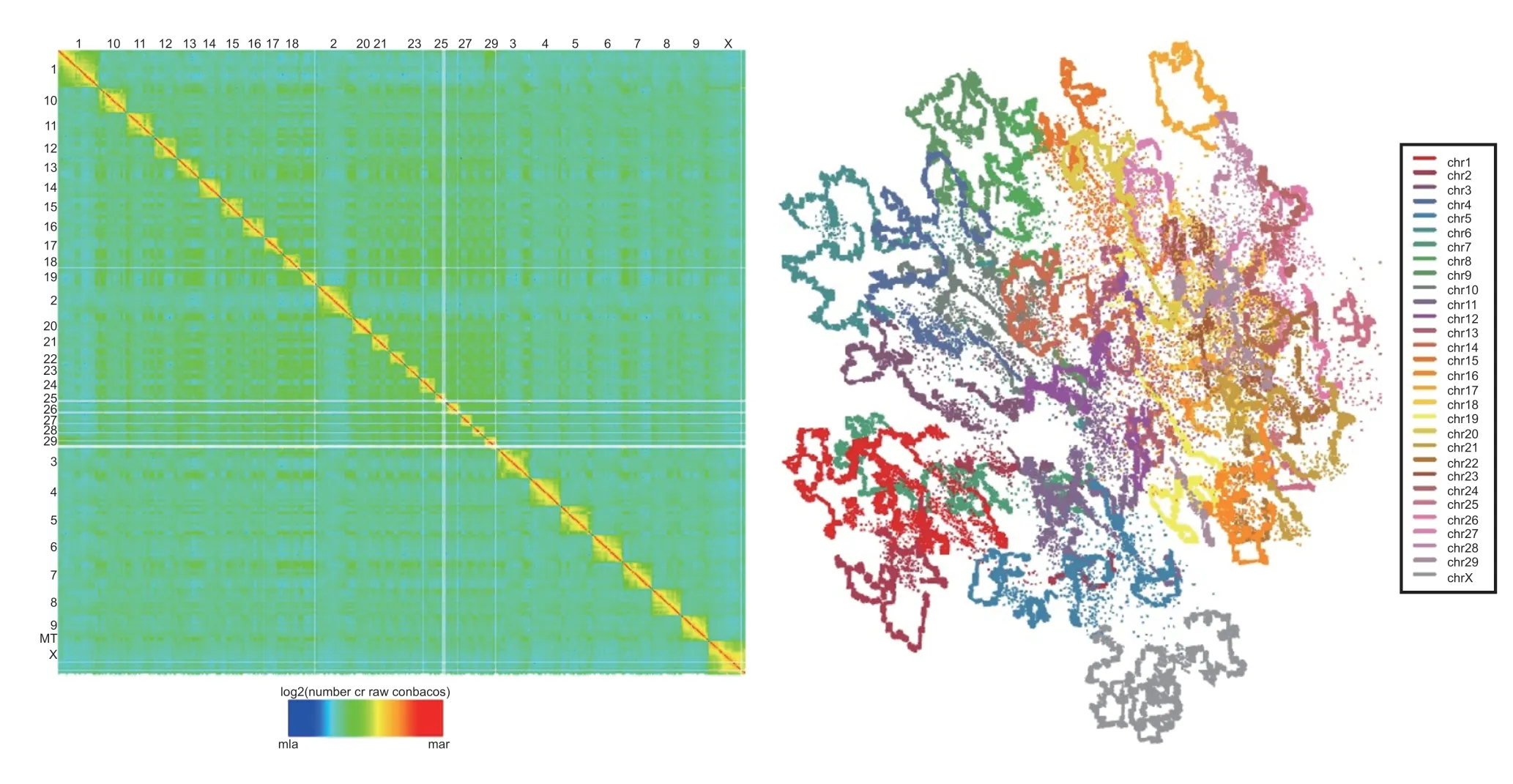
Fig.2 Overview of the Hi-C heatmap matrix (left) and a simulated 3D genome structure (right) in cattle skeletal muscle.In the Hi-C heatmap matrix (left),horizontal and vertical coordinates are chromosome numbers.Each chromosome is represented by a different color in the simulated structure (right).

Fig.3 3D genome transcriptional control.Transcriptional upregulation of TOMD1 during the conversion of bovine fetal into adult muscle tissue is accompanied by loop formation(purple curve) in the 3D genome structure’s TADs (yellow triangle) and compartments(reddish brown area),involving novel loops and increased accessible chromatin by ATACseq and H3K27ac and H3K4me1 enhancer signatures in genomic regions.A1,adult 1;A2,adult 2;F1,fetal 1;F2,fetal 2.
GWAS hits,expression quantitative trait loci (eQTL),and selection signatures have been examined for regulatory elements to provide a more comprehensive atlas of associations between gene regulation and livestock traits.GWAS has been used to identify genomic loci associated with livestock traits,which are significantly enriched for regulatory elements (Xiangetal.2019).Combined with eQTL,GWAS signals influence genes expressed in trait-relevant tissues in cattle (Fangetal.2020;Prowse-Wilkinsetal.2021;Liu Setal.2022).Selection signatures,which are signatures left on the genome by long-term natural or artificial selection,have also been examined to determine if regulatory elements are under selection.Promoters and enhancers have been found to be significantly enriched in genomic selection signals in sheep,highlighting the importance of gene regulatory elements in the domestication process (Naval-Sanchezetal.2018).Combining the information from GWAS,eQTL,and selection signatures in the study of regulatory elements may provide a more comprehensive understanding of the relationships between gene regulation and livestock traits and aid in the identification of causal mutations for molecular breeding.
Currently,the Functional Annotation of Animal Genomes Consortium (FAANG) is working to create reference functional maps for farmed animals.It is profiling their transcription and chromatin accessibility landscapes to deepen our understanding of the features and functions of the noncoding genome and improve the precision and sensitivity of animal breeding.It has shown that some key mutations located in ruminant regulatory elements,such as two candidate variations in the pleiomorphic adenoma gene zinc finger 1 (PLAG1) gene’s intergenic region (ss319607405 [14:23375648-23375650] and ss319607406 [14: 23375692]),are associated with cattle stature (Karimetal.2011).In addition,a 504-bp deletion~14 kb downstream of the fibroblast growth factor 5 (FGF5) gene affects its enhancer activity and is associated with goat villus (Caietal.2020).Similarly,information about mutations in the functional elements of livestock has been deposited in RGD v2.0,constituting a major update of the ruminant functional and evolutionary genomics database (http://animal.nwsuaf.edu.cn/) (Fuetal.2021).
Beneficial mutations could be transferred between populations or species or designed at will.A growing list of putative target genes and mutations for genome editing has been or is being pursued to improve livestock production (Laibleetal.2015;Lievensetal.2015;Van Eenennaam 2017),regarding characteristics such as growth rate,muscle mass,and meat fat content.Combining gene editing and 3D genome mapping data may redefine key mutations or genomic regions associated with various trait variants.While editing key genes during development may cause embryonic death,editing key regulatory elements may only improve such traits without lethality.
In addition,DNA loops can be created to mediate enhancer-promoter interactions at specific genomic loci using CRISPR-based approaches (Morganetal.2017;Kimetal.2019).Studying their roles in transcription control could open the door to a new world in animal breeding.Forced chromatin looping may ultimately enable the 3D genome to be equipped with the potential to improve livestock quality.For example,enhancers could be removed from genes that adversely affect livestock growth and development or closed into genes that can improve livestock quality.Therefore,we can manipulate DNA loop formation to enhance beneficial gene expression and reduce harmful gene expression to speed up the breeding process without damaging the genome sequence.
There are substantial advancements in 3D genome research of humans and model animals.The study of the 3D genome provides valuable insights into the molecular mechanisms of complex traits,such as revealing causal mutations in GWAS and explaining the effects of structural or copy number variations on phenotypic variations by annotating TAD boundaries.However,research on livestock remains in its infancy,primarily focusing on the identification of genome structures and regulatory elements that could be utilized in the future by integrating them with animal breeding techniques,such as work on the targets of genome selection (GS) or gene editing,to improve animal breeding quality.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (31972558) and the Agricultural Improved Seed Project of Shandong Province,China(2020LZGC014).
Declaration of competing interest
The authors declare that they have no conflict of interest.
Ethical approvalThe Institutional Animal Care and Use Committee (IACUC)of the Northwest A&F University,China has approved the use of animals and all experimental protocols of this study.
杂志排行

Journal of Integrative Agriculture的其它文章
- Advances in DNA methylation and its role in cytoplasmic male sterility in higher plants
- Assessment of molecular markers and marker-assisted selection for drought tolerance in barley (Hordeum vulgare L.)
- SUPER WOMAN 2 (SPW2) maintains organ identity in spikelets by inhibiting the expression of floral homeotic genes OsMADS3,OsMADS58,OsMADS13,and DROOPING LEAF
- Genetic dissection and validation of a major QTL for grain weight on chromosome 3B in bread wheat (Triticum aestivum L.)
- Coupling of reduced inorganic fertilizer with plant-based organic fertilizer as a promising fertilizer management strategy for colored rice in tropical regions
- Effect of chemical regulators on the recovery of leaf physiology,dry matter accumulation and translocation,and yield-related characteristics in winter wheat following dry-hot wind