内质网应激相关基因能预测骨肉瘤预后并与肿瘤免疫微环境相关
2024-01-13周逸驰
张 丹,周逸驰
(1.武汉大学中南医院,武汉 430000;2.武汉市第四医院 脊柱二骨肿瘤科,武汉 430000)
骨肉瘤起源于间叶组织,是一种侵袭性恶性肿瘤,高发于儿童与青少年,具有病情进展迅速、易转移、易复发、预后不佳等特点[1-2],约有30%患者在确诊5年后出现进展性转移[3]。尽管立体定向放射治疗、碳离子放射治疗、标准化学治疗、手术切除等综合性治疗方案近年来有了较大发展,但骨肉瘤患者的5年生存率仍不理想[4]。内质网( Endoplasmic reticulum, ER )参与蛋白质合成、加工与转运,钙离子的储存以及脂质和类固醇激素的合成与转运等多种生理活动。当细胞受到各种理化因素刺激时,内质网功能的内稳态体系被打破,由正常状态转变为应激状态,称为内质网应激(Endoplasmic reticulum stress, ERS ) 。ERS主要表现为ER腔内错误折叠与未折叠蛋白大量沉积、钙离子平衡失调等;细胞通过激活未折叠蛋白反应(Unfold protein response, UPR)等信号通路引导和参与ER中异常蛋白质的再折叠和降解,防止细胞功能进一步受损。然而持续或过强的ERS则会诱导细胞凋亡信号通路的启动。多种研究表明内质网应激与肿瘤的发生及进展有关,本研究基于内质网应激相关基因以生物信息学方法构建了骨肉瘤患者的风险模型,并探索了其与肿瘤免疫微环境的关系。
1 资料与方法
1.1 数据下载
训练集的转录组数据及相应的临床信息下载于UCSC Xena数据库;验证集的转录组数据与临床信息下载于GEO数据库(GSE21257,GSE39058),两组数据使用R软件包“inSilicoMerging”[5]进行了合并,随后使用R软件包“limma”的removeBatchEffect函数对合并后的数据去除批次效应。在GeneCards网站(www.genecards.org)搜索条目“endoplasmic reticulum stress”,并提取relevance score≥7.0的基因作为内质网应激相关基因。
1.2 风险模型的构建与验证
使用R软件包“survival”评估了每个基因的预后显著性,提取logrankP<0.05的基因;随后使用维恩图(Jvenn)[6]对预后相关基因与ERS相关基因取交集。使用R软件包“glmnet”的LASSO-cox方法提取了风险特征基因,此外还设置了5折交叉验证以获得最优模型。对上述获得的基因使用R软件包“survival”进行多因素COX回归分析,最终筛选P<0.05的风险特征基因构建风险模型。风险评分=β1×expression of gene 1 +β2×expression of gene 2 +β3×expression of gene 3 + … +βn×expression of gene n 。使用五种临床特征(种族、性别、年龄、转移状态及原发肿瘤部位)对风险评分进行分组并进行比较以验证风险模型的独立性。
1.3 风险模型与临床特征的相关性
使用R软件包“rms”建立了列线图,并评估了生存时间、生存状态、风险评分、临床特征在样本中的预后显著性。随后使用了R软件包“survival”和“stdca.R”进行3、5年的决策曲线分析(Decision curve analysis, DCA)以评估该风险模型的临床效用;最后评估了风险评分与5种临床特征(种族、性别、年龄、转移状态及肿瘤原发部位)之间的关系。
1.4 生存分析
使用R软件包“survival”与“survminer”以Kaplan-Meier生存曲线评估了高、低风险组之间预后差异的显著性;随后用R软件包“pROC”的roc函数进行了1、3、5年的受试者工作特征曲线(Receiver operating characteristic curve, ROC)分析,并使用ci函数评估曲线下面积(Area under curve, AUC)和置信区间;随后使用R软件包“pheatmap”绘制了风险预后热图,显示不同风险组样本的生存状态分布及风险特征在不同分组之间的表达情况。
1.5 免疫分析
使用ESTIMATE算法[7]评估了高低风险组之间免疫评分、基质评分和ESTIMATE评分之间的差别;使用ssGSEA方法[8]比较了28种不同的免疫细胞在高低风险分组样本之间的浸润丰度。随后使用了MCP-counter算法[9]评估了8种不同的免疫细胞及两种基质细胞(血管内皮细胞与纤维母细胞)在高低风险分组之间的浸润丰度;并使用相关性散点图评估了风险评分与免疫评分、基质评分及ESTIMATE评分的相关性。
1.6 功能富集分析
使用R软件包“limma”[10]对高、低风险组之间进行了基因差异表达分析,随后使用Metascape[11]对差异表达基因(Differentially expressed genes, DEGs)进行了进一步的分析。使用了R软件包“clusterProfiler”[12]对DEGs进行了基因本体论(Gene ontology, GO)与京都基因与基因组百科全书(Kyoto encyclopedia of genes and genomes, KEGG)联合功能富集分析,“GOplot”包[13]计算每个条目的zscore并对结果进行可视化。基于分子特征数据库 (http://www.gsea-msigdb.org/gsea/downloads.jsp) 下载了GO生物过程基因集以评估相关途径及分子机制,使用R软件包“GSVA”[14]计算了每个样本在每个基因集中的富集得分。基于MSigDB数据库,使用R软件包“clusterProfiler”包进行了基因集富集分析(Gene set enrich analysis, GSEA)。
1.7 统计学分析
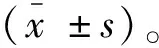
2 结果
2.1 构建ERS相关风险模型
从UCSC Xena数据库(https://xenabrowser.net/datapages/)下载了88例患者的临床信息与相应基因表达数据作为训练集,其中3例无随访信息予以剔除;GEO数据库(https://www.ncbi.nlm.nih.gov/geo/,GSE21257,GSE39058)共下载95例患者信息与相应基因表达数据作为验证集,训练集与验证集临床数据详见表1。训练集数据使用单因素COX回归分析显示共有7 714个基因与骨肉瘤预后相关,将该预后相关基因集与内质网应激相关基因集(n=770)取交集,提取出79个内质网应激-预后相关基因(见图1a),这79个基因在人类染色体的坐标、表达情况及相互联系如图1b所示。随后使用LASSO回归分析(LASSO regression algorithm)进一步提取出26个候选基因,最佳λ值选择为0.041 4(见图1c-1d)。基于这26个候选基因,采用多因素COX回归分析最终确定了6个风险特征基因:TMED10、VEGFA、MAPK10、TOR1B、PTGIS、SERPINH1,其中VEGFA、PTGIS及SERPINH1与骨肉瘤患者的不良预后相关,而TMED10、MAPK10及TOR1B与骨肉瘤患者的良好预后相关(见图1e)。风险模型构建公式:风险评分= 0.239×VEGFA表达量-2.45362×TMED10表达量-0.94029×MAPK10表达量-1.45051×TOR1B表达量+ 0.79041×PTGIS表达量+ 1.03296×SERPINH1表达量。ROC分析显示(见图1f),该风险模型1年ROC曲线下面积(Area under curve, AUC)为0.95(95%CI:1.00-0.88),3年AUC为0.97(95%CI:1.00-.094),5年AUC为0.98(95%CI: 1.00-0.95)。训练集决策曲线分析(Decision curve analysis, DCA)显示,风险评分的预测的准确性高于其余临床预测指标(年龄、性别、种族、原发肿瘤部位及转移状态,见图1g)。以风险评分的平均值将训练集患者分为高风险组(n=43)与低风险组(n=42),K-M生存曲线显示,低风险组预后显著优于高风险组(Log-rankP<0.001,见图1h)。

表1 训练集与验证集的临床数据特征Table 1 Clinical data characteristics of the training and validation sets
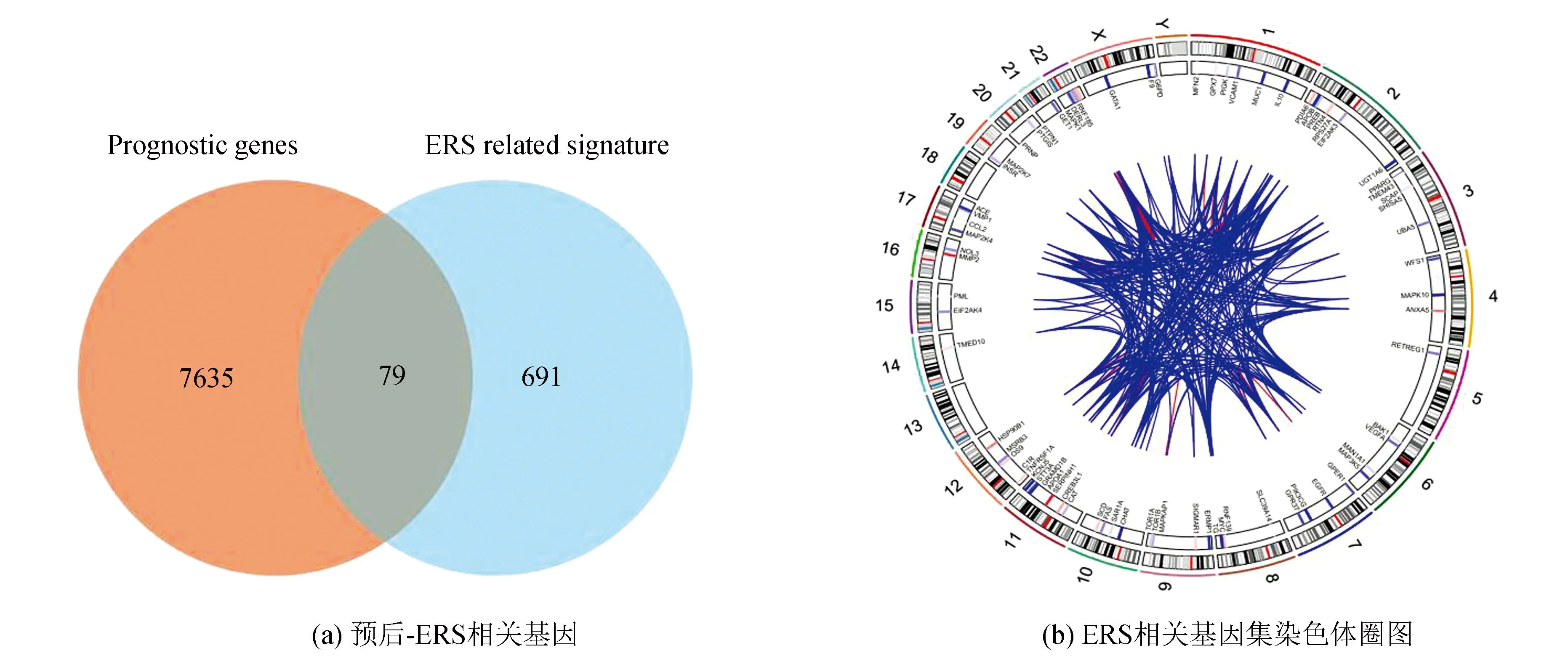

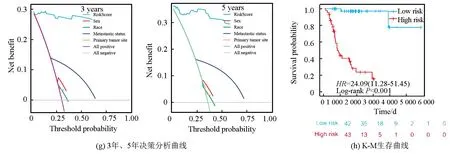
图1 训练集数据建立ERS基因相关风险模型Fig.1 Establishment of the ERS genes related risk model in the training cohort
2.2 基因差异表达分析与富集分析
训练集中,对高、低风险组进行基因表达差异分析(设定差异倍数为1.5,显著阈值为0.05),共有341个差异表达基因(Differential expressed genes, DEGs),其中有138个上调基因,203个下调基因(见图2a)。Metascape分析显示,DGEs共分为6个模块,其主要富集于淋巴细胞介导的免疫力、细胞外基质组成、骨化、凋亡程序的调控及应对缺氧(见图2b,2c)。GO-KEGG联合富集分析结果显示,DEGs主要富集于循环免疫球蛋白介导的体液免疫反应、补体激活的经典途径、免疫球蛋白复合体、金色葡萄球菌感染(见图2d)。GSVA结果显示,肥大细胞活化参与的免疫反应、成熟B细胞分化参与的免疫反应、淋巴细胞活化参与的免疫反应、T细胞活化参与的免疫反应、B细胞活化参与的免疫反应、巨噬细胞活化参与的免疫反应、免疫反应激活富集于低风险组,而缺氧诱导因子1 α信号通路、对缺氧反应的内在凋亡信号通路、细胞对缺氧反应的调节、凋亡染色体凝缩、翻译后蛋白质靶向内质网膜、内质网未折叠蛋白质反应富集于高风险组(见图2e)。GSEA分析显示,DEGs主要富集于获得性免疫反应、先天性免疫反应、淋巴细胞与非淋巴细胞间的免疫调节作用及补体系统(见图2f)。上述分析表明,与免疫细胞激活相关的信号通路主要富集于低风险组,而低风险组预后优于高风险组,表明低风险组的免疫景观及免疫状态可能优于高风险组;同时,与内质网过度应激的相关通路及缺氧反应的相关通路富集于高风险组,提示内质网应激及缺氧反应可能为导致骨肉瘤患者预后不佳的原因。
2.3 风险模型的独立性
本研究基于五种临床特征验证了该风险模型的独立性,种族(P=0.55)、性别(P=0.4)、年龄(P=0.5)、转移状态(P=0.16)及原发肿瘤部位(P=0.99、P=0.88、P=0.74)之间的风险评分均无统计学差异(见图3a)。随后,以性别(见图3b,女性P<0.001,男性P<0.001)、转移状态(见图3c,转移P=0.002,非转移P<0.001)、年龄(见图3d,18岁以下P<0.001,18岁以上P=0.013)及种族(见图3e,白种人P<0.001,非白种人P<0.001)分组进行生存分析(见图3b),该风险模型仍旧显示出了强大的预后预测能力,低风险组预后均显著优于高风险组。

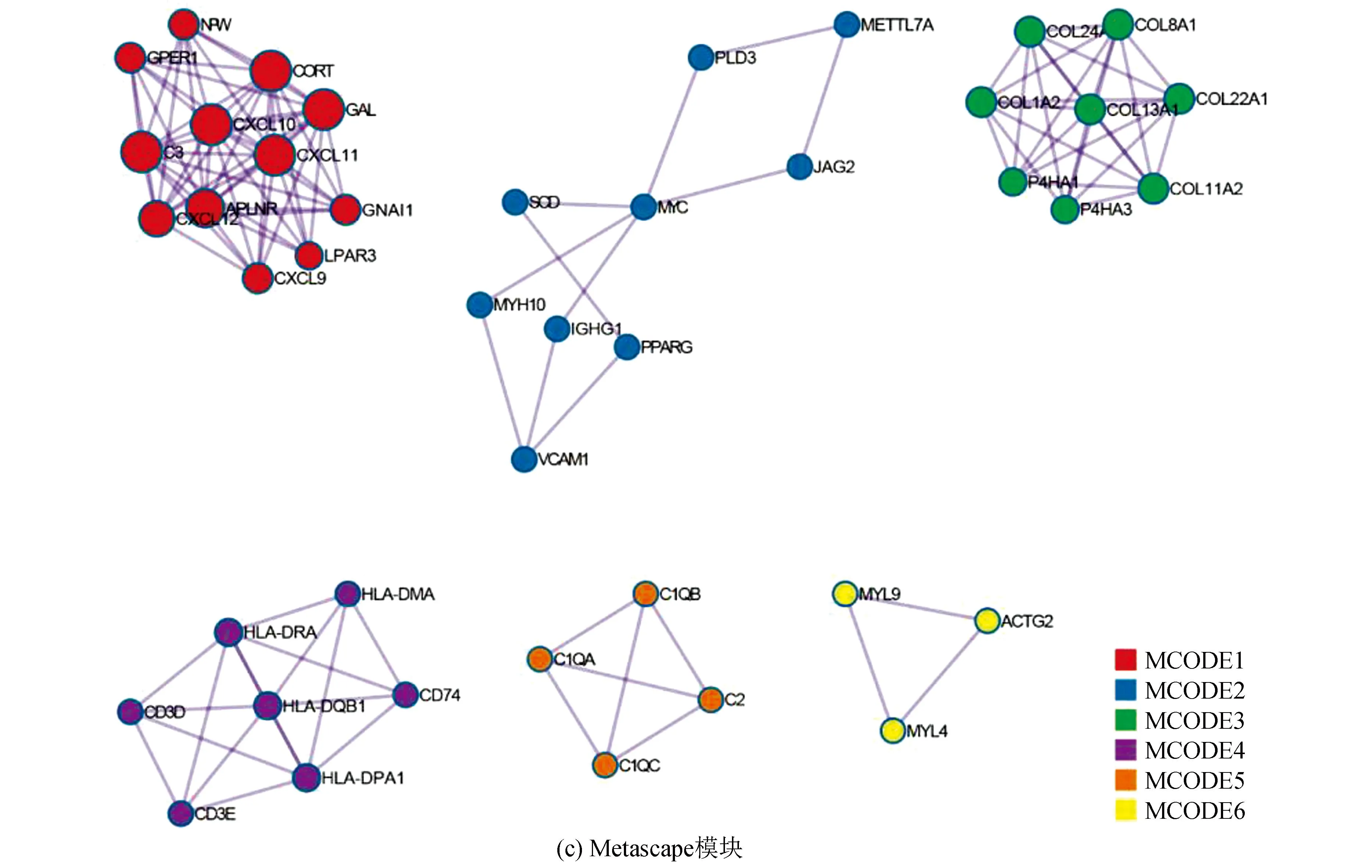
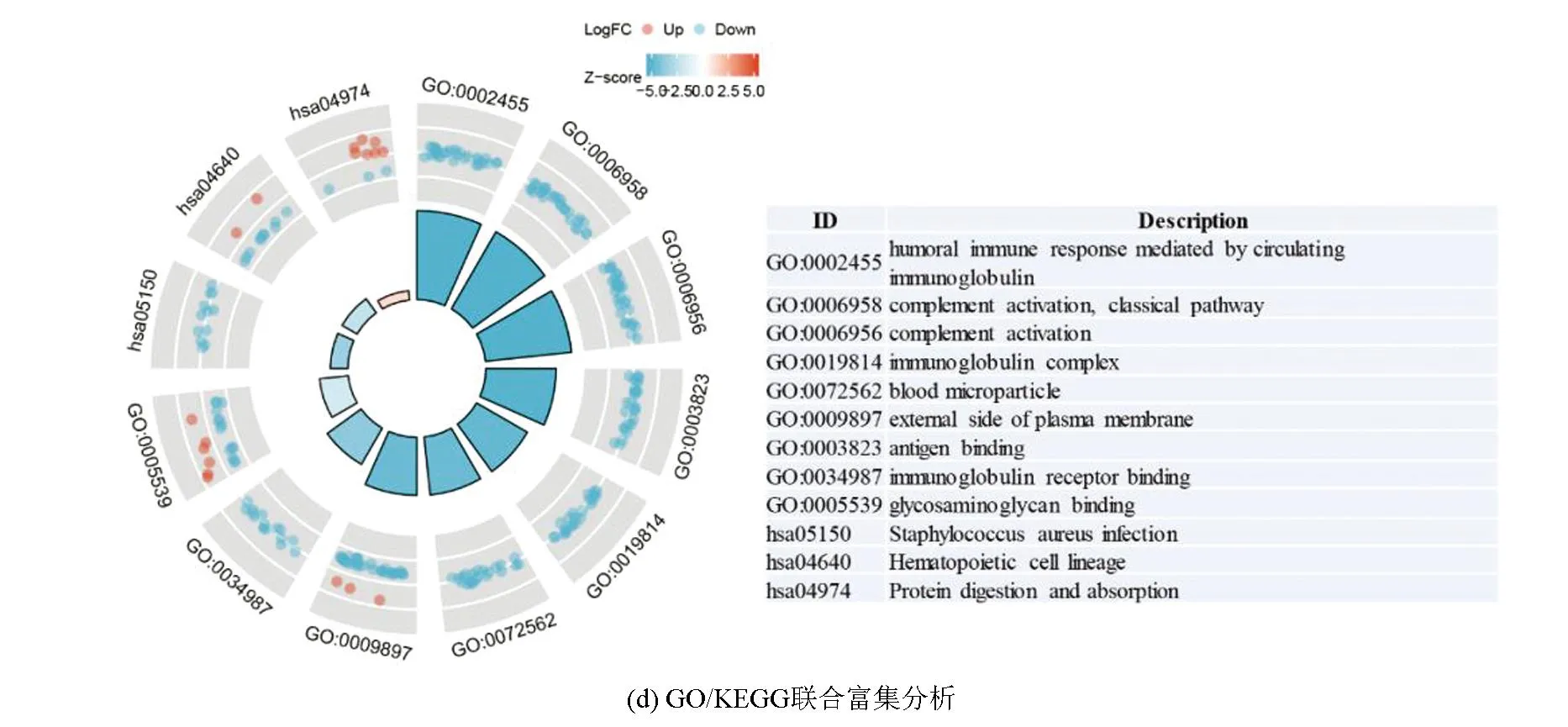

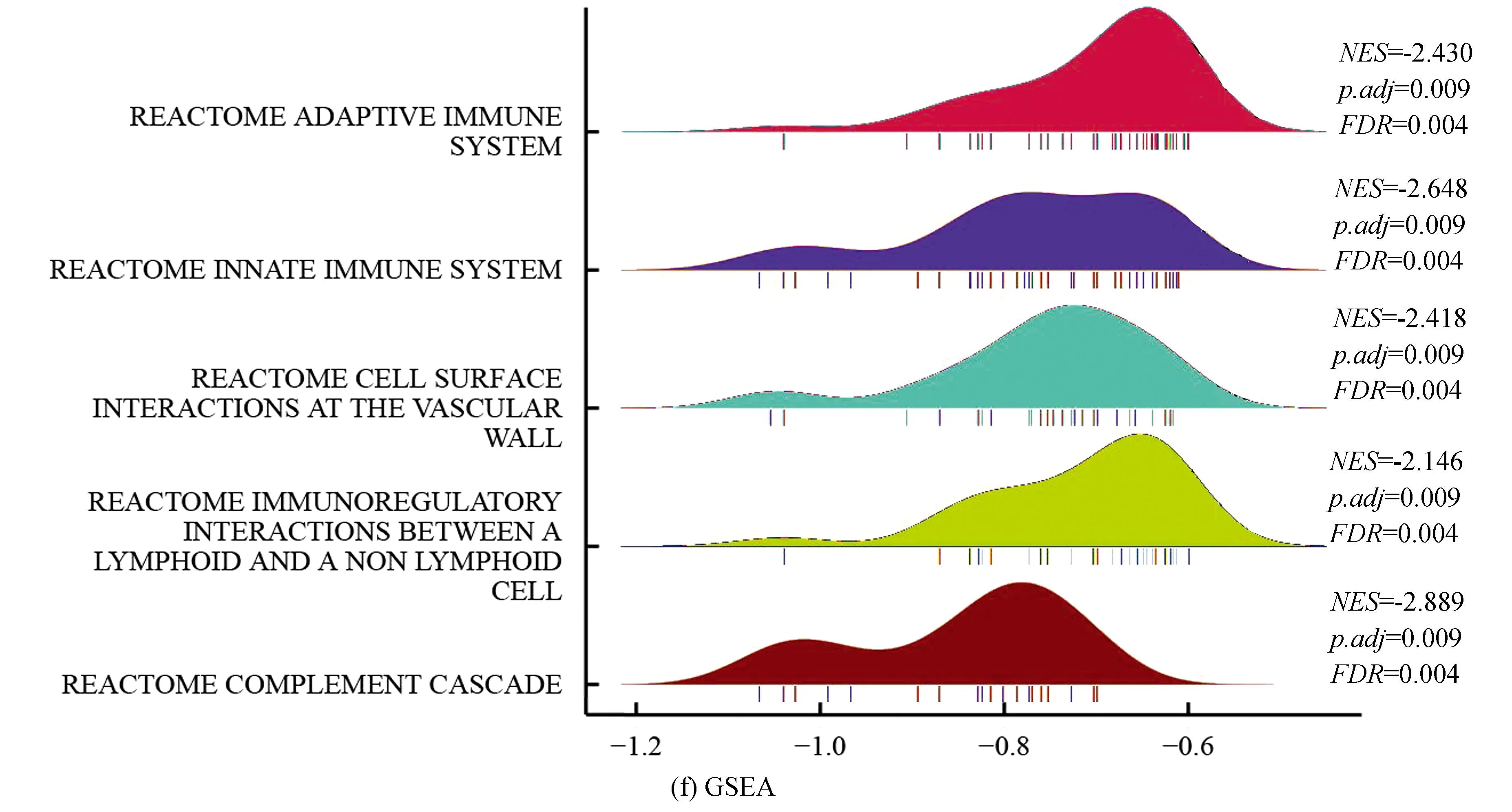
图2 高低风险组DEGs相关分析Fig.2 DEGs related analysis of the high risk and low risk group

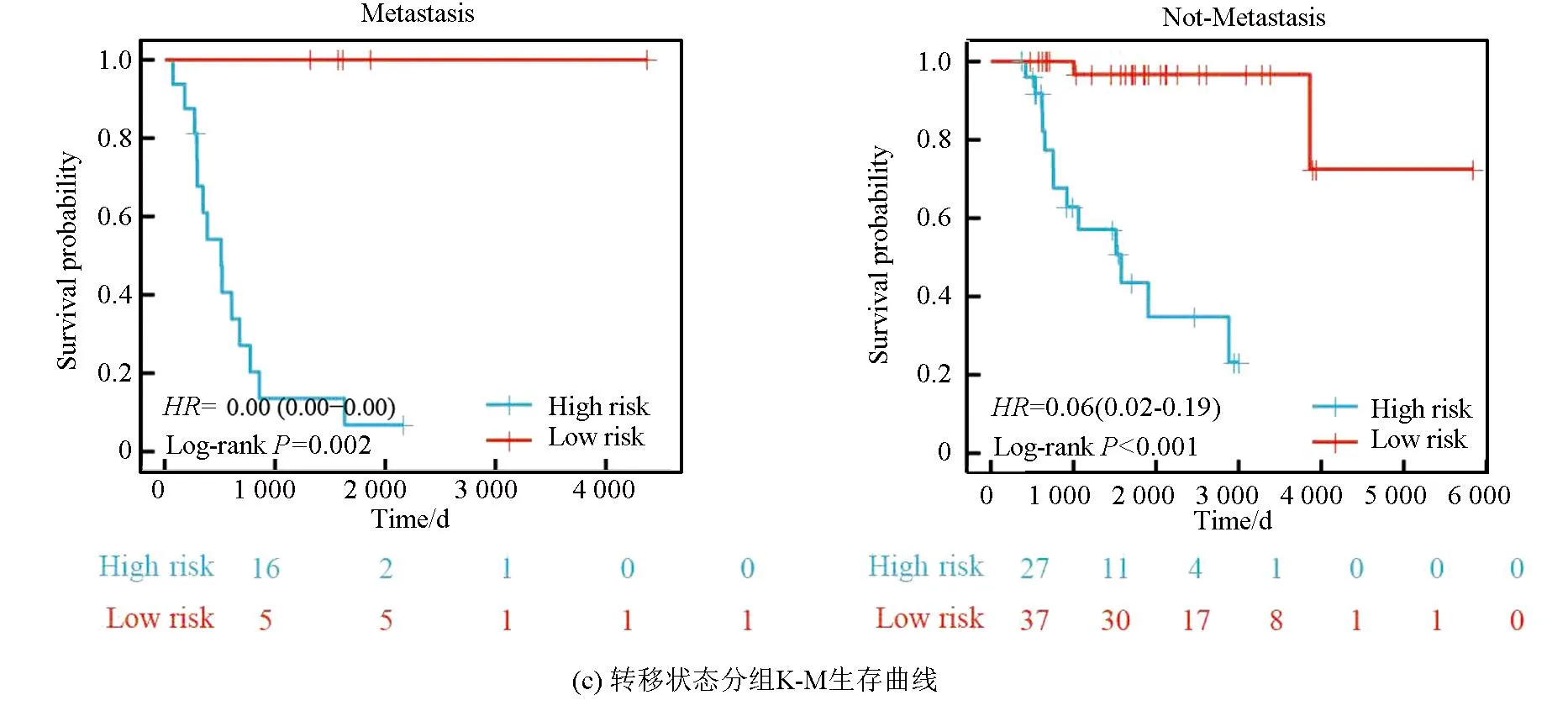
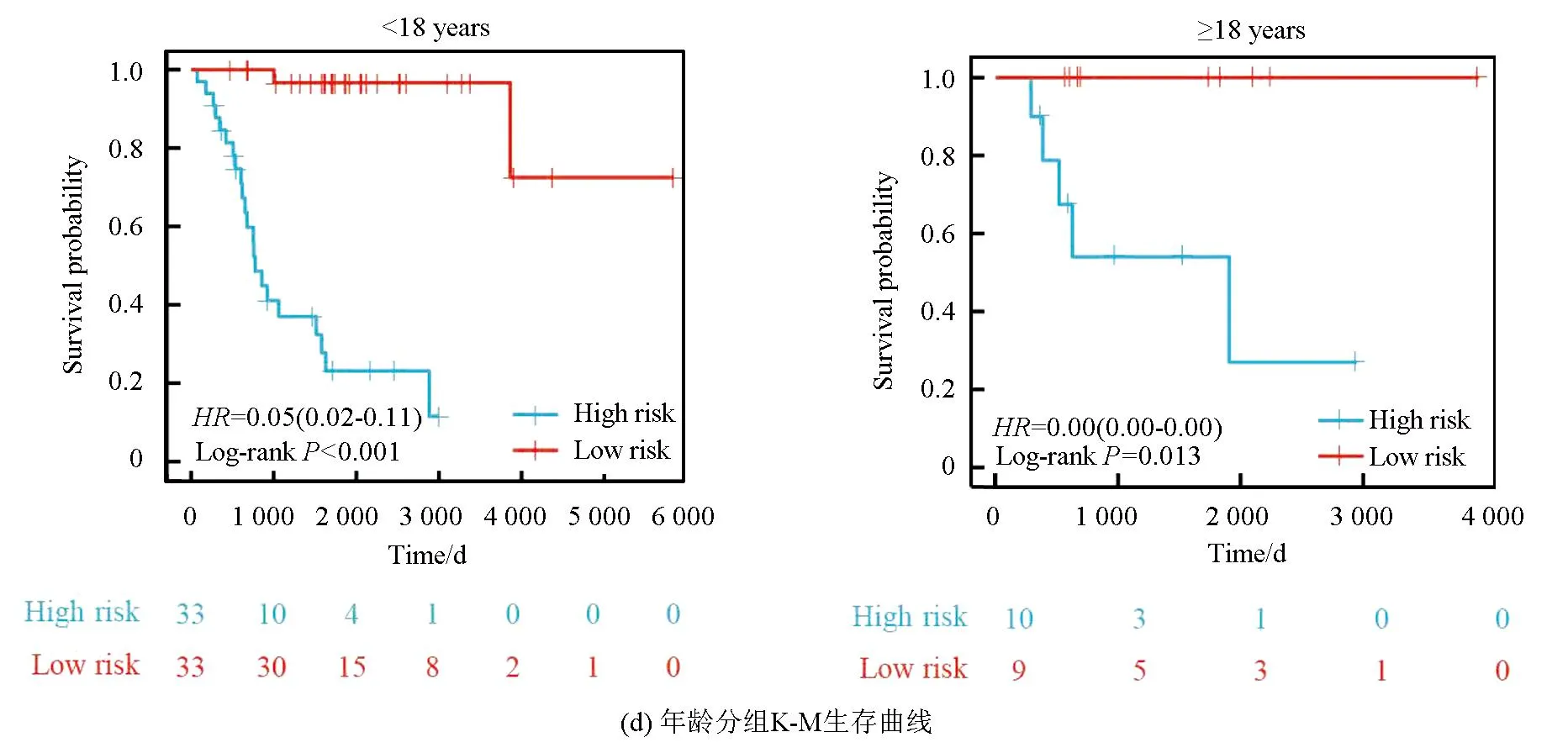
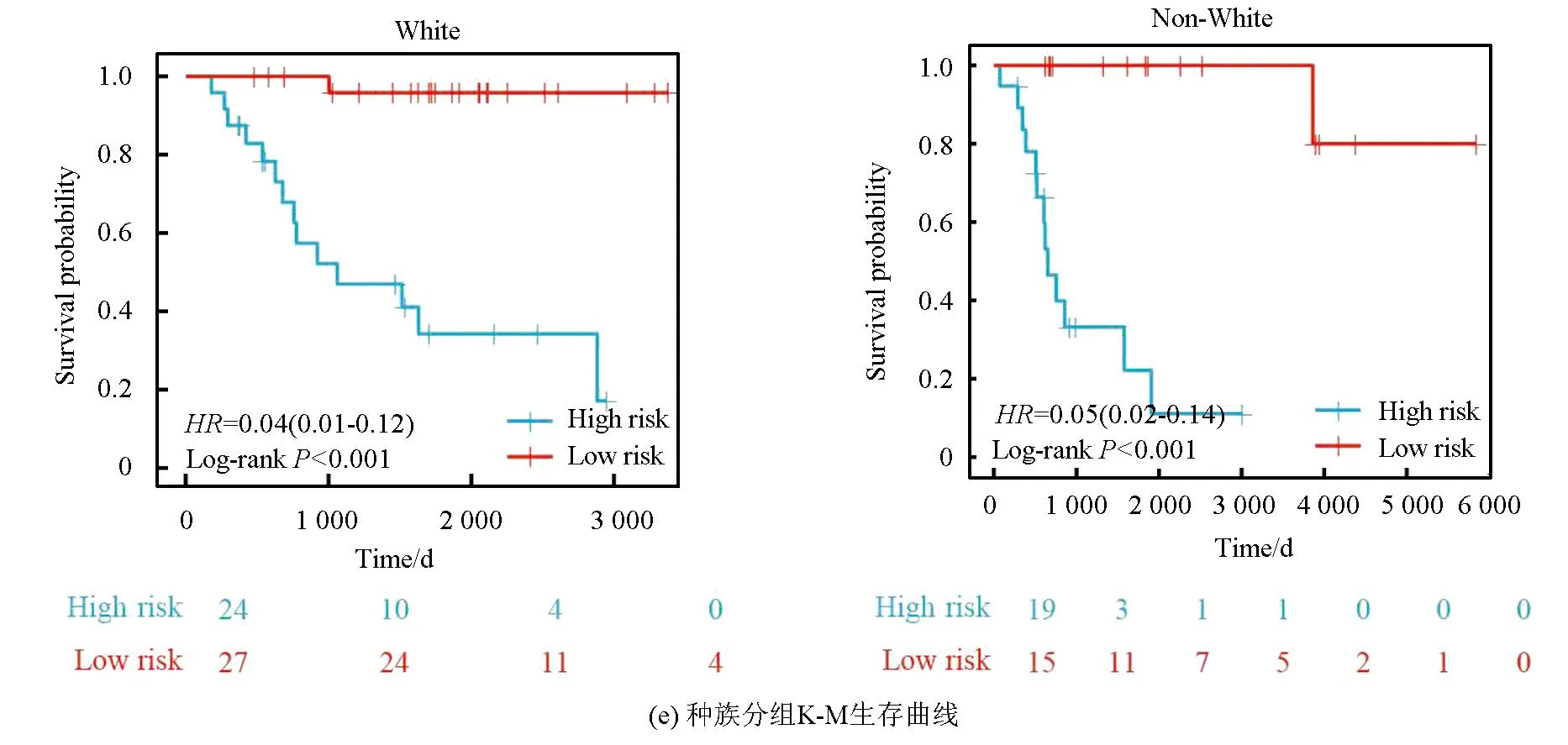
图3 风险模型独立性评估Fig.3 Independence assessment of the risk model
2.4 列线图的构建
整合风险评分与临床特征(性别、种族、原发肿瘤部位、转移状态)构建了列线图以更加准确地预测骨肉瘤患者的预后(见图4a),根据风险评分与上述临床特征对骨肉瘤预后的影响,给予相应的评分,随后分别在训练集与验证集中以校准曲线验证了该列线图结果,校准曲线显示了该列线图良好的预后能力(见图4b,4c)。模型总体C指数为0.967 3(95%CI:0.944 8-0.989 8),P<0.001。
2.5 风险特征基因的特点
图5a表明,该风险模型成功将训练集的骨肉瘤患者分为高风险组与低风险组,在高风险组中VEGFA、PTGIS及SERPINHI高表达,而在低风险组中TMED10、MAPK10及TOR1B高表达。图5b表明,VEGFA、PTGIS及SERPINHI在高风险组中的表达量高于低风险组(P<0.05),而TMED10、MAPK10及TOR1B在低风险组中的表达量高于高风险组(P<0.05)。生存分析(见图5c)显示,TMED10、MAPK10及TOR1B的高表达与良好预后相关(Log-rankP<0.05),而VEGFA、PTGIS及SERPINHI的高表达与不良预后相关(Log-rankP<0.05)。
2.6 免疫分析
ESTIMATE分析显示,训练集的低风险组的基质评分、免疫评分及ESTIMATE评分均高于高风险组(P<0.05),进一步表明低风险组与免疫反应相关(见图6a)。相关性散点图分析表明,风险评分与基质评分(P<0.001,R=-0.39),免疫评分(P<0.05,R=-0.26)及ESTIMATE评分(P<0.001,R=-0.365)呈负相关(见图6b)。MCP-counter分析(见图6c)显示,T细胞、CD8 T细胞、B细胞系、NK细胞、单核细胞系及内皮细胞在低风险组中的浸润丰度高于高风险组(P<0.05)。ssGSEA分析(见图6d,6e)显示,28种不同的免疫细胞主要富集于低风险组,其中活化B细胞、活化CD8 T细胞、记忆B细胞、1型辅助T细胞、2型辅助T细胞、髓源性抑制细胞、NK细胞及NK T细胞的富集评分有统计学差异(P<0.05)。上述结果表明低风险组具有较高的免疫景观与免疫评分,而高风险组则具有较低的免疫景观与免疫评分。

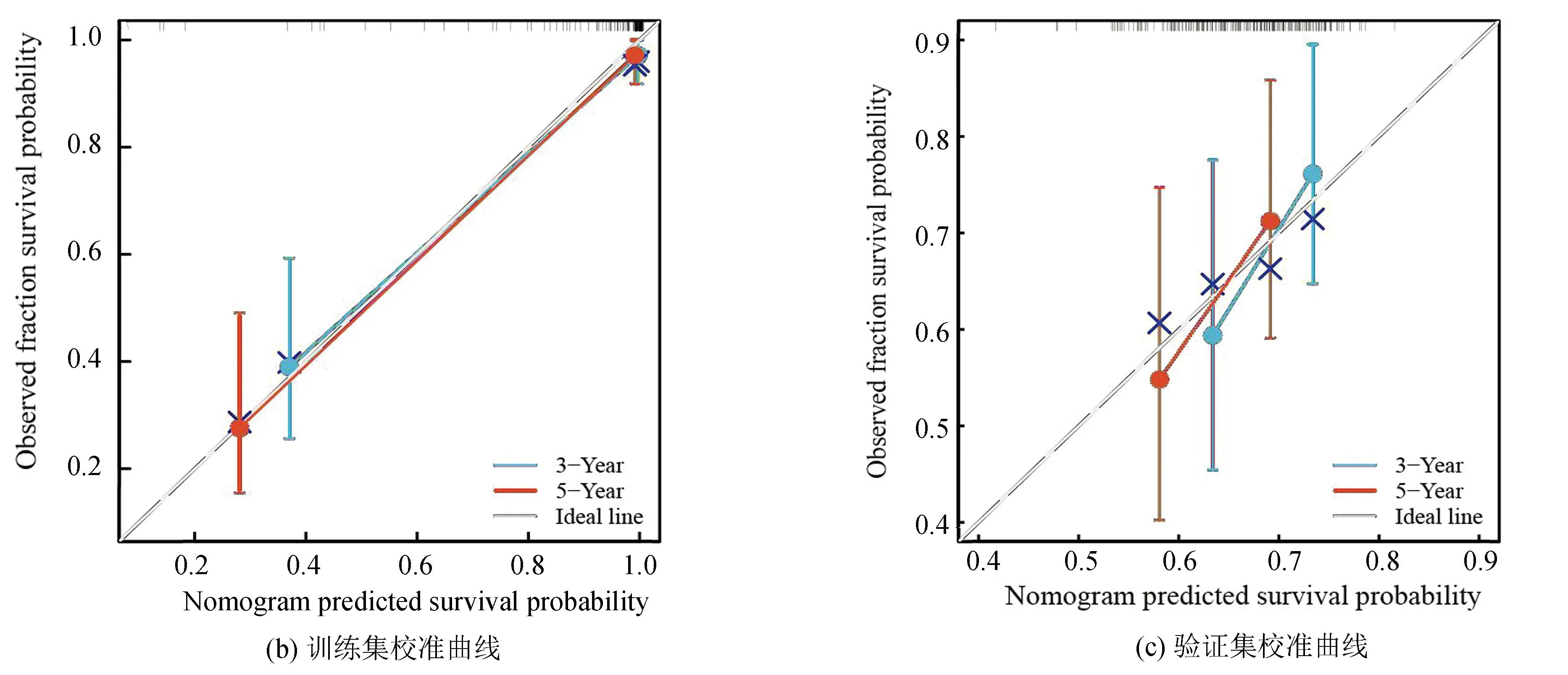
图4 构建列线图与校准曲线Fig.4 Establishment of nomograph and calibration curve
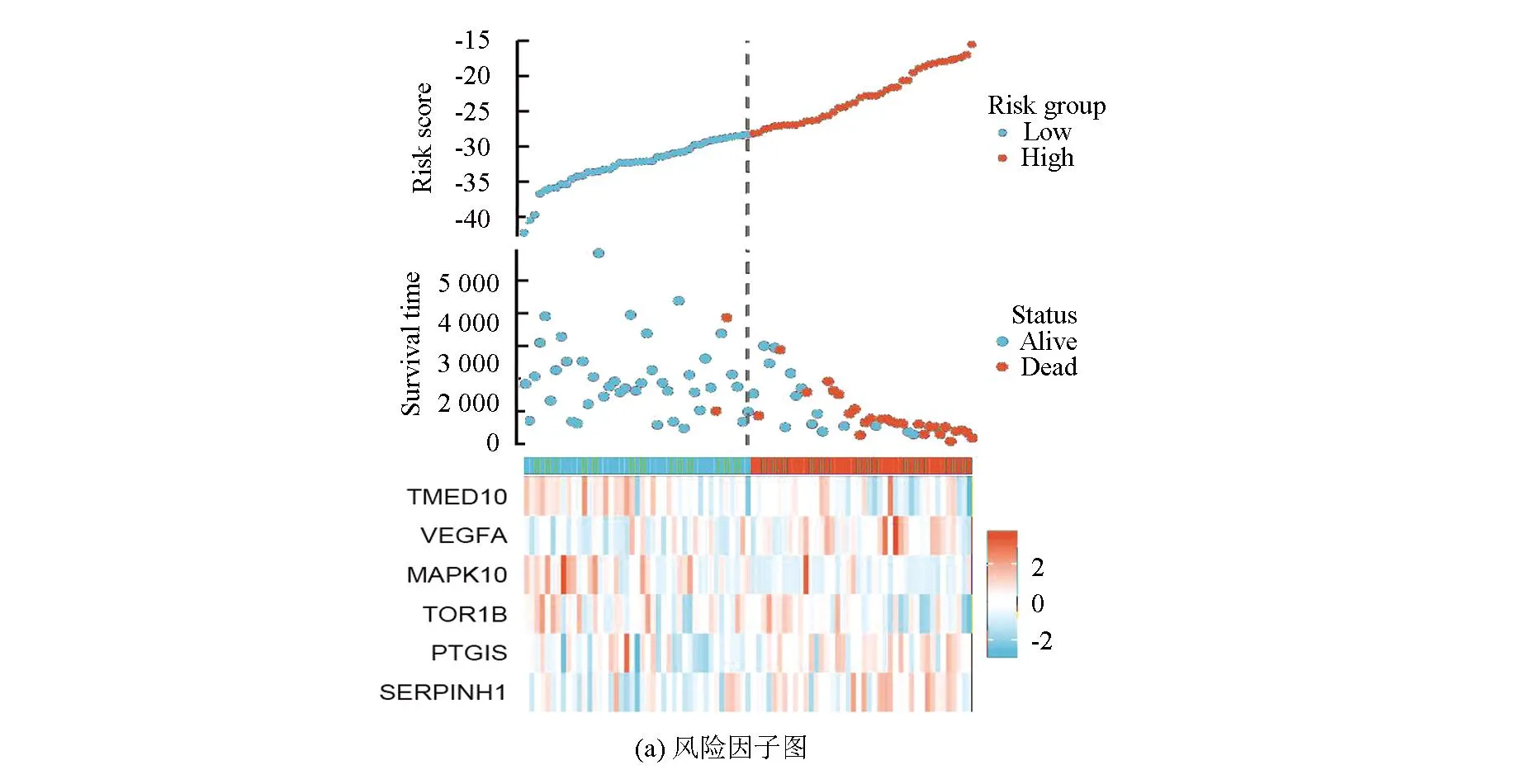
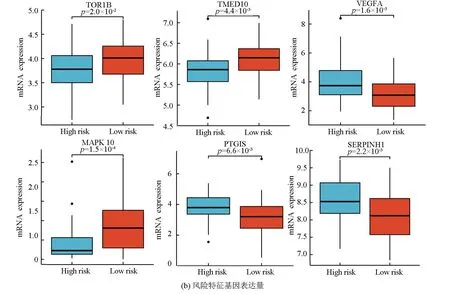
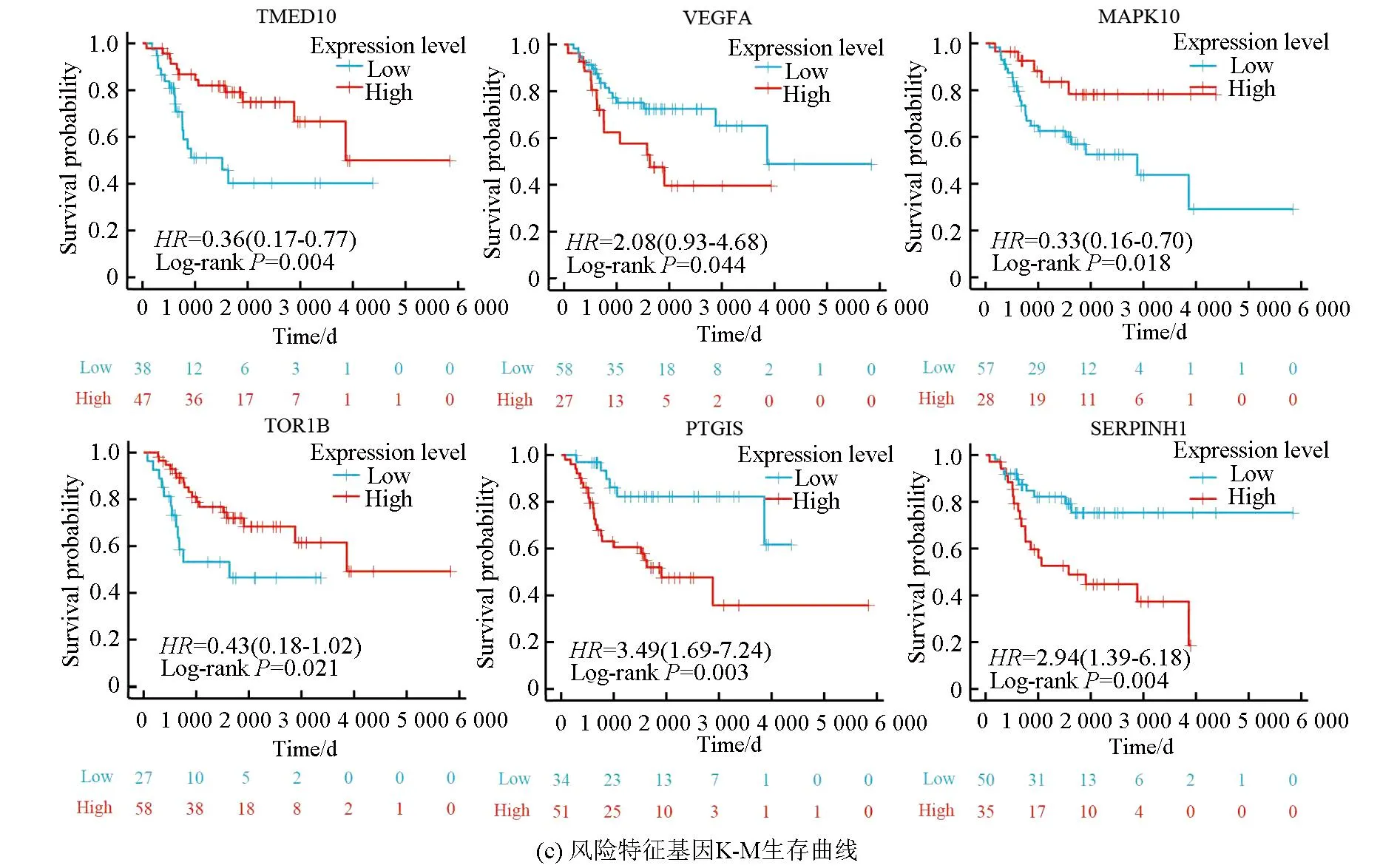
图5 风险特征基因的特点Fig.5 Characteristics of risk genes
2.7 验证风险模型
在验证集中,风险评分同样将骨肉瘤患者分为高风险组与低风险组,热图可见VEGFA、PTGIS及SERPINH1在高风险组高表达,而TMED10、MAPK10及TOR1B在低风险组高表达(见图7a)。图7b表明,VEGFA、PTGIS及SERPINH1在高风险组表达高于低风险组(P<0.05),而TMED10、MAPK10及TOR1B在低风险组表达高于高风险组(P<0.05)。图7c表明低风险组预后优于高风险组。ROC分析显示(见图7d),1年AUC为0.59(95%CI: 0.72-0.45),3年AUC为0.63(95%CI: 0.76-0.49),5年AUC为0.62(95%CI: 0.77-0.48),显示该预后模型在验证集中也有较好的预测能力。上述分析结果与训练集一致,进一步表明该风险模型具有优良的预测能力。
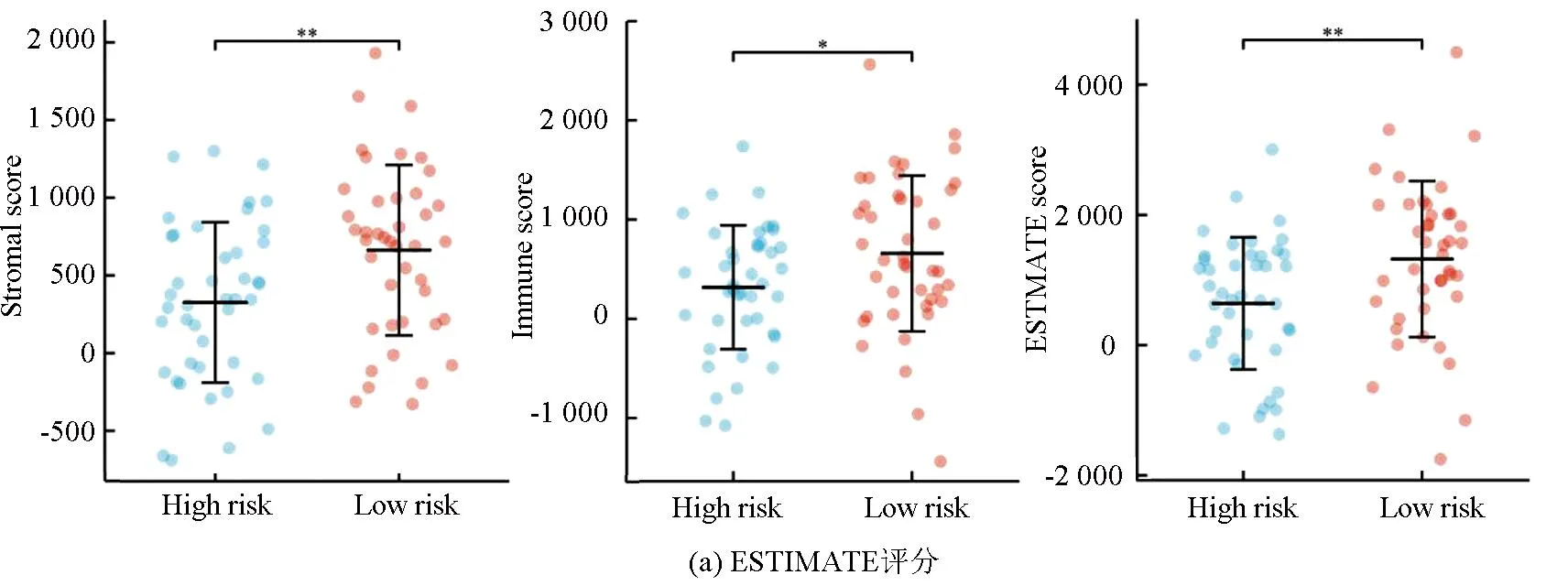
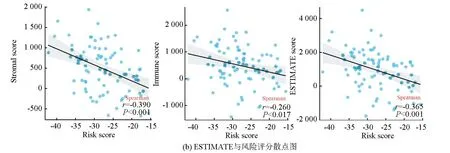

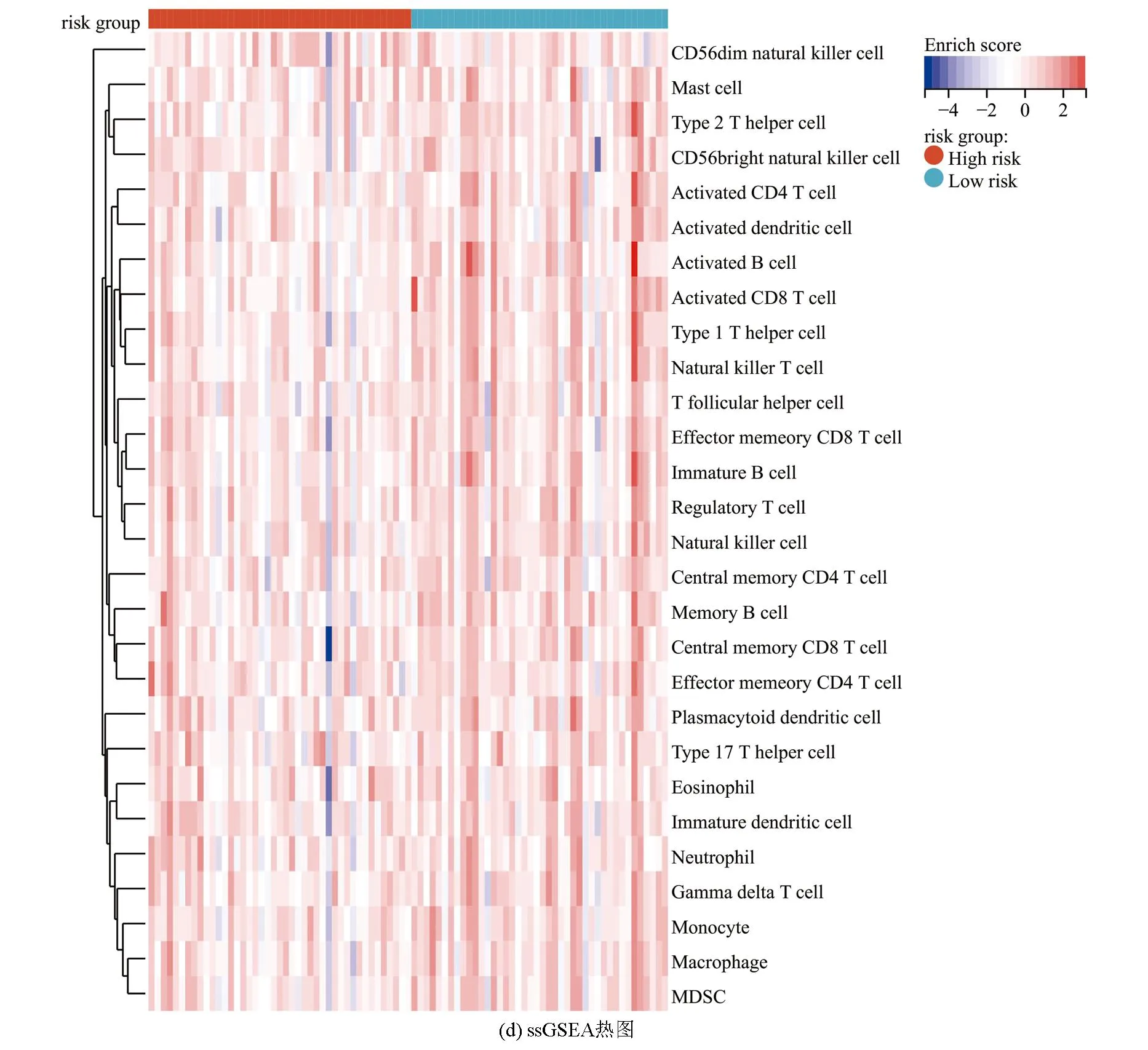
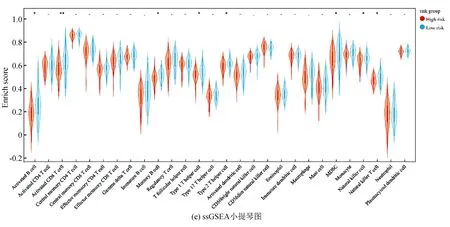
图6 免疫分析Fig.6 Immune analysis

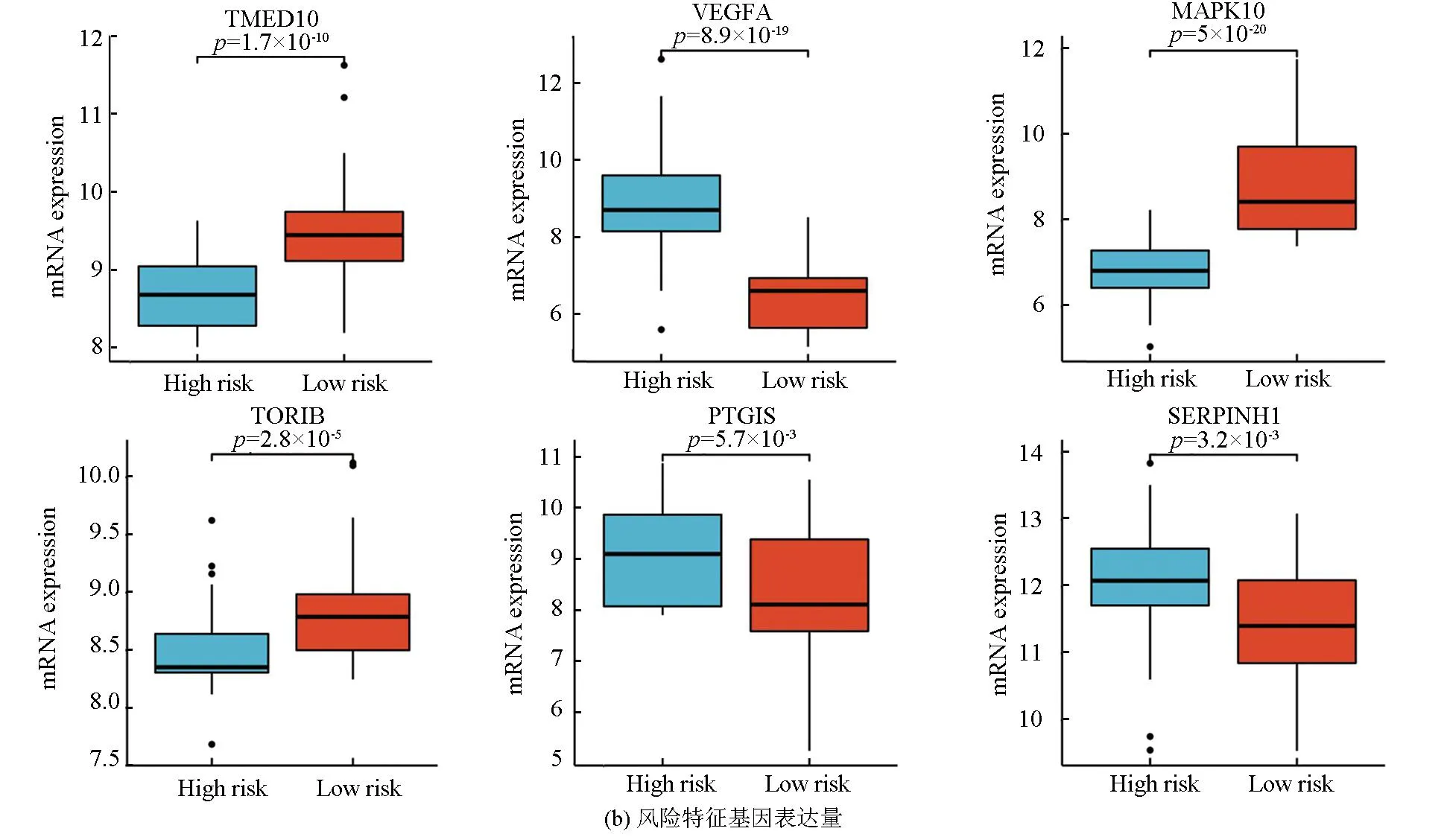
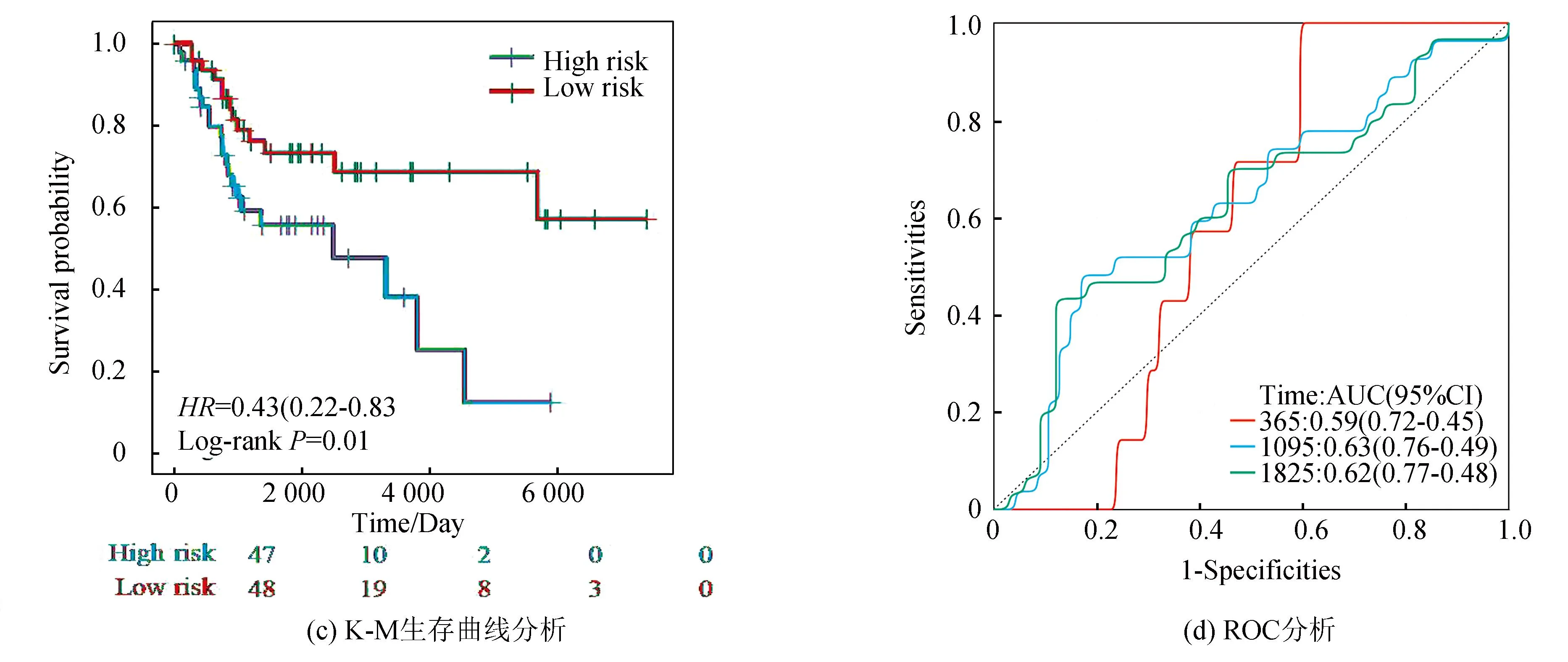
图7 验证风险模型的鲁棒性Fig.7 Verification of the robustness of the risk model
3 讨 论
骨肉瘤是儿童和青少年最常见的恶性骨肿瘤[15],尽管近年来综合治疗方案快速发展,但骨肉瘤的5年生存率仍不理想,目前迫切需要制定有效的风险分层方法[16-17]。内质网应激(Endoplasmic reticulum stress,ERS)在人类癌症领域中的作用得到了广泛关注,为骨肉瘤的个体化疗法带来了新的希望。内质网是蛋白质合成和细胞内钙储存最重要的细胞器,并参与多种细胞信号通路的调控[18-19]。在细胞内和细胞外应激诱导下,未折叠或错误折叠蛋白的积累可激活未折叠蛋白反应(Unfolded protein response, UPR),从而恢复内质网的内环境平衡[20]。PERK/ATF4/CHOP、IRE-1/XBP1和ATF6信号通路在调控ERS诱导的细胞生理反应中起重要作用[21-22],然而过度、持续的内质网应激激活也可诱导细胞凋亡[23]。同时,ERS参与了多种疾病的进程,如帕金森病、阿尔兹海默症、糖尿病及肿瘤[24-26],对ERS的深入研究可为骨肉瘤的临床治疗提供重要参考。在恶性肿瘤中,癌细胞不断暴露于缺氧的肿瘤微环境和细胞内DNA损伤应激中,ERS激活的风险较高,PERK-ATF4分支可激活上皮-间充质转化(Epithelial-To-mesenchymal transition, EMT)反应,促进乳腺癌细胞转移[27]。在肝细胞癌和前列腺癌中,来自UPR的转录因子可以结合并激活VEGF的启动子,导致内皮细胞的增殖和迁移[28-29]。此外,ERS激活也能诱导癌细胞休眠, 细胞休眠通过将癌细胞阻滞在细胞周期的G0/G1期,从而阻断癌细胞的分裂和增殖[30],处于静止状态生存的癌细胞对放化疗不敏感,最终导致肿瘤复发,患者预后不良 。此外,相关研究证明IRE-1-XBP1信号通路可以抑制抗肿瘤免疫应答,为肿瘤的形成提供机会[31]。上述结论提示ERS可能在肿瘤免疫微环境(Tumor immune microenvironment, TIME)的重构中起重要作用,是免疫治疗的潜在靶点。虽然多项研究证实了ERS在骨肉瘤中的治疗价值,但ERS在骨肉瘤中的生物学功能尚未完全了解,特别是其在调节TIME的作用机制。
本研究基于UCSC Xena数据库及GeneCards数据库以生物信息学方法构建了一个ERS基因相关骨肉瘤风险模型,该风险模型包括6个风险特征基因:P24跨膜转运蛋白10(Transmembrane P24 trafficking protein 10, TMED10)、血管内皮生成因子A(Vascular endothelial growth factor A, VEGFA)、丝裂原激活的蛋白激酶(Mitogen-activated protein kinase, MAPK10)、Torsin家族1成员B(Torsin family 1 member B, TOR1B)、前列腺素I2合酶(Prostaglandin I2 synthase, PTGIS)及丝氨酸蛋白酶抑制剂1(Serine proteinase inhibitor, SERPINH1),其中TMED10、MAPK10及TOR1B与骨肉瘤患者的良好预后相关,而VEGFA、PTGIS及SERPINH1与骨肉瘤患者的不良预后相关。MAPK10是肝细胞肝癌(Hepatocellular carcinoma, HCC)的重要抑癌基因,其表达丢失提示HCC患者的预后较差[32],另外MAPK10低表达也与宫颈癌患者的不良预后相关[33];VEGFA在多种肿瘤组织中高表达,不仅与肿瘤血管生成相关,还能直接或间接参加肿瘤免疫反应[34];PTGIS高表达可促进肿瘤相关巨噬细胞(Tumor-associated macrophages, TAMs)和调节性T细胞(T-regulatory cells, Tregs)在肿瘤微环境中的浸润,并与肺癌、卵巢癌及胃癌患者的不良预后有关[35],另外PTGIS过表达也能作为结直肠癌肝转移的预测指标[36];SERPINH1是多种肿瘤不良预后的危险因素和预测因子,同时其过表达与肿瘤免疫抑制状态有关,SERPINH1可作为泛癌免疫治疗的潜在靶点[37-38]。本研究的结果表明ERS过度激活与恶性临床病理特征和基因组改变密切相关,同时这些结果揭示了ERS在骨肉瘤中的意义及其影响ERS激活的分子机制。该风险模型将骨肉瘤患者分为了高风险组与低风险组,生存分析表明低风险组预后优于高风险组。同时ROC分析、DCA均表明该风险模型能准确地预测骨肉瘤患者预后,随后进一步验证了该风险模型的独立性。
本研究对高、低风险组患者的基因进行差异分析,并对DEGs进行富集分析,结果揭示了应对缺氧、ERS等相关通路富集于高风险组,而免疫反应相关通路富集于低风险组。TIME在肿瘤患者预后方面具有重要作用,因为肿瘤的发展与周围基质的修饰有关,而免疫细胞是肿瘤基质的关键组成部分[39]。ESTIMATE算法可以根据基因表达值推断肿瘤纯度,以及肿瘤中免疫细胞和基质细胞的比例[7],因此能够准确地反应TIME。本研究结果表明,具有高免疫评分、基质评分及ESTIMATE评分的骨肉瘤患者具有良好预后。另外,本研究还采用了另外两种方法—MCP counter算法及ssGSEA来评估高低风险组肿瘤组织中的免疫细胞浸润丰度。MCP counter算法结果表明高风险组中有5/8的不同免疫细胞丰度显著降低,与ESTIMATE结果一致,表明免疫景观(Immune landscape)在高风险组下调。ssGSEA分析概述了28种免疫相关细胞的丰度,结果表明高风险组患者处于相对较低的免疫状态,进一步证实了ESTIMATE和MCP counter的结果。在肿瘤微环境中,ERS在肿瘤的免疫机制中也发挥了重要作用,其不仅决定了肿瘤细胞的命运,而且还间接触发了免疫抑制反应,例如ERS的中性粒细胞通过凝集素样氧化低密度脂蛋白受体-1(Lectin-like oxidized low-density lipoprotein receptor-1,LOX-1)的表达获得免疫抑制活性[40],口腔鳞状细胞癌的ERS可传递给中性粒细胞,导致中性粒细胞上调其表面受体LOX-1并导致其免疫功能受到抑制[41],综上所述,缺氧、过度ERS等肿瘤免疫微环境的变化可能引起了高风险组患者的免疫抑制状态,而免疫反应的激活可能与低风险组患者的良好预后相关。
虽然已有学者从肿瘤微环境、免疫细胞浸润和能量代谢等方面构建骨肉瘤风险模型[42-44],但本研究仍具有独特的优点:(1)本研究聚焦于ERS,构建了由6个风险特征基因组成的风险模型,并将骨肉瘤患者分为了具有显著生存差异的高、低风险组;(2)本研究探索了不同分组之间的DEGs,并进一步探索了DEGs的富集通路,结果表明这些通路与免疫反应有关;(3)本研究验证了该风险模型的准确性与独立性,并进一步在GEO队列中验证了该风险模型;第四,本研究探索了这六种风险特征基因的特点,其结果在训练集与验证集中一致;最后,免疫分析表明高风险组具有较低的免疫景观及免疫评分,推测过度ERS可能导致免疫抑制状态,从而引起高风险组的不良预后。
4 结 论
以生物信息学方法挖掘公共数据库,筛选出6个内质网应激相关风险特征基因,并以此为基础构建了骨肉瘤患者的风险模型,为骨肉瘤患者的预后提供了新思路、新方法,具有一定的新颖性;本研究所得到的基因可作为骨肉瘤基础研究和治疗的潜在靶点,有一定的应用价值;不足之处在于本研究仍具有一定的局限性:一方面,本研究结果来源于生物信息学分析,并没有得到进一步的实验验证;另一方面,本研究使用的数据从开放数据库下载获得的,且回顾性研究的低证据水平的性质仍然存在,还需要更多的前瞻性研究来进一步证实ERS在骨肉瘤中的预后价值;最后,关于该风险模型与骨肉瘤患者免疫状态的关系需要进一步的实验验证。
