肥厚型心肌病家系中MYBPC3-D1149fs*40新发突变的基因型及临床表型研究
2024-01-11陆晓晨曹鑫南许培曙陈雨枫盛红专
陆晓晨,曹鑫南,许培曙,刘 虎,顾 漪,陈雨枫,盛红专
(南通大学附属医院心血管内科,南通大学医学院,江苏 南通 226001)
肥厚型心肌病(hypertrophic cardiomyopathy,HCM)是常染色体显性遗传性心脏病,主要病因是编码肌小节蛋白或肌小节相关结构蛋白的基因变异,目前已发现至少30个基因中超过2 000种突变与HCM有关[1],其中肌球蛋白结合蛋白C(myosin binding protein C3,MYBPC3)和β-肌球蛋白重链(β-myosin heavy chain,MYH7)占50%以上[1-2]。不同HCM基因突变导致的临床表型具有显著异质性[3],识别与家族性HCM相关的基因突变、分析基因型与表型之间的关联对于疾病的诊断和防治具有重要意义。本研究纳入1例HCM患者及其家庭成员,取外周血行全外显子测序,发现了一种新的MYBPC3突变位点——c.3445(exon31)delG.p.Asp1149fs*40,携带该突变的患者均表现为非梗阻性肥厚型心肌病。
1 资料与方法
1.1 一般资料
本研究纳入2023年2月就诊于南通大学附属医院心内科的1例HCM患者及其家庭成员,HCM纳入标准及排除标准参照《中国成人肥厚型心肌病诊断与治疗指南2023》。采集HCM患者及家庭成员的临床及心电图和超声心动图资料。本研究经我院伦理委员会批准实施(批号:2019-K021-01),所有研究对象均签署知情同意书。
1.2 基因测序
采用乙二胺四乙酸(EDTA)抗凝管抽取先证者及其家系成员外周静脉血各2 mL,提取DNA样本进行质检。质检合格后由北京智因基因公司捕获先证者的外显子序列并完成全外显子测序。
结果对照人类参考基因组进行变异分析,并通过生物信息分析软件预测变异的有害性。查找数据库(Gene、Genome和OMIM等)和相关文献检索筛选出候选致病基因及突变位点。
2 结 果
2.1 基因测序结果
先证者第31号外显子上的3 445位碱基G缺失,导致第1 149位天冬氨酸的编码序列开始出现移码突变,在移码后生成40个氨基酸提前引入终止密码子从而结束转录翻译过程,导致肽链缩短无法完整表达蛋白,见图1。该突变位点在dbSNP、千人基因组计划等数据库中均未见报道。进一步对该家系成员进行测序验证,发现该家系中另2例HCM患者均携带相同突变,其余4例表型阴性成员则无此突变,见图2。

图2 MYBPC3基因c.3445(exon 31)delG的家系图谱
2.2 突变氨基酸种间保守性分析和致病性预测
利用clustalx软件分析发现,上述突变氨基酸在不同物种间高度保守,见图3A。使用SIFT、Polyphen_2等生物信息学软件预测该突变位点的致病性,4种软件预测的结果一致,提示MYBPC3-D1149fs*40的突变位点为有害突变,见图3B。
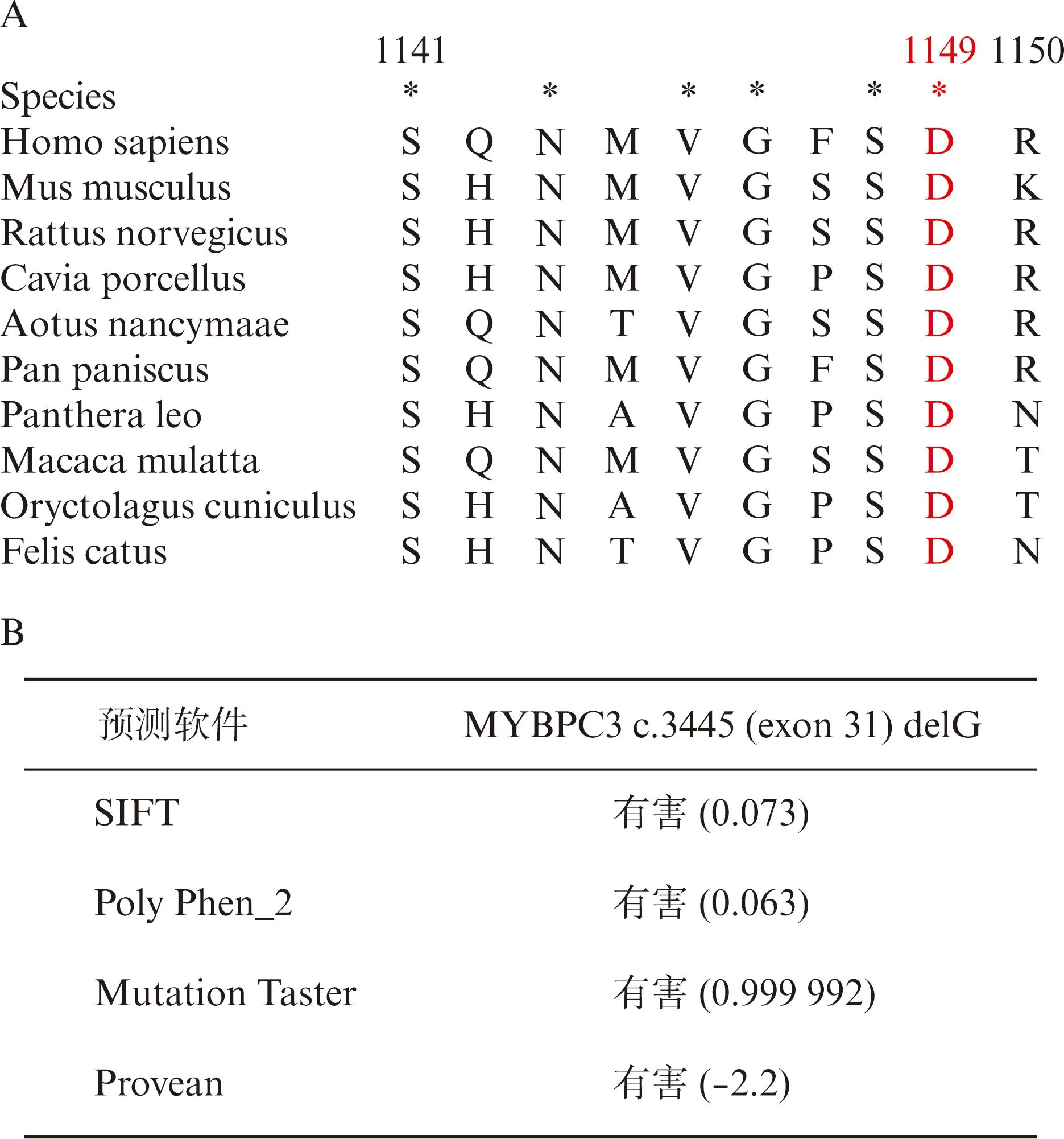
图3 MYBPC3基因c.3445(exon 31)delG.p.Asp1149fs*40突变
2.3 该家系成员临床表型和基因型
该家系中先证者(Ⅱ-1)为74岁女性,因“劳累后胸闷气短10年”就诊。3年前心电图提示窦性心律、房性早搏、左心室肥大、ST-T改变,见图4A;超声心动图显示不对称室间隔增厚(室间隔厚度17.0 mm,左心室后壁厚度11.0 mm),左心室射血分数66.0%,见图4B。该患者无高血压、瓣膜病等疾病,临床考虑肥厚型心肌病。
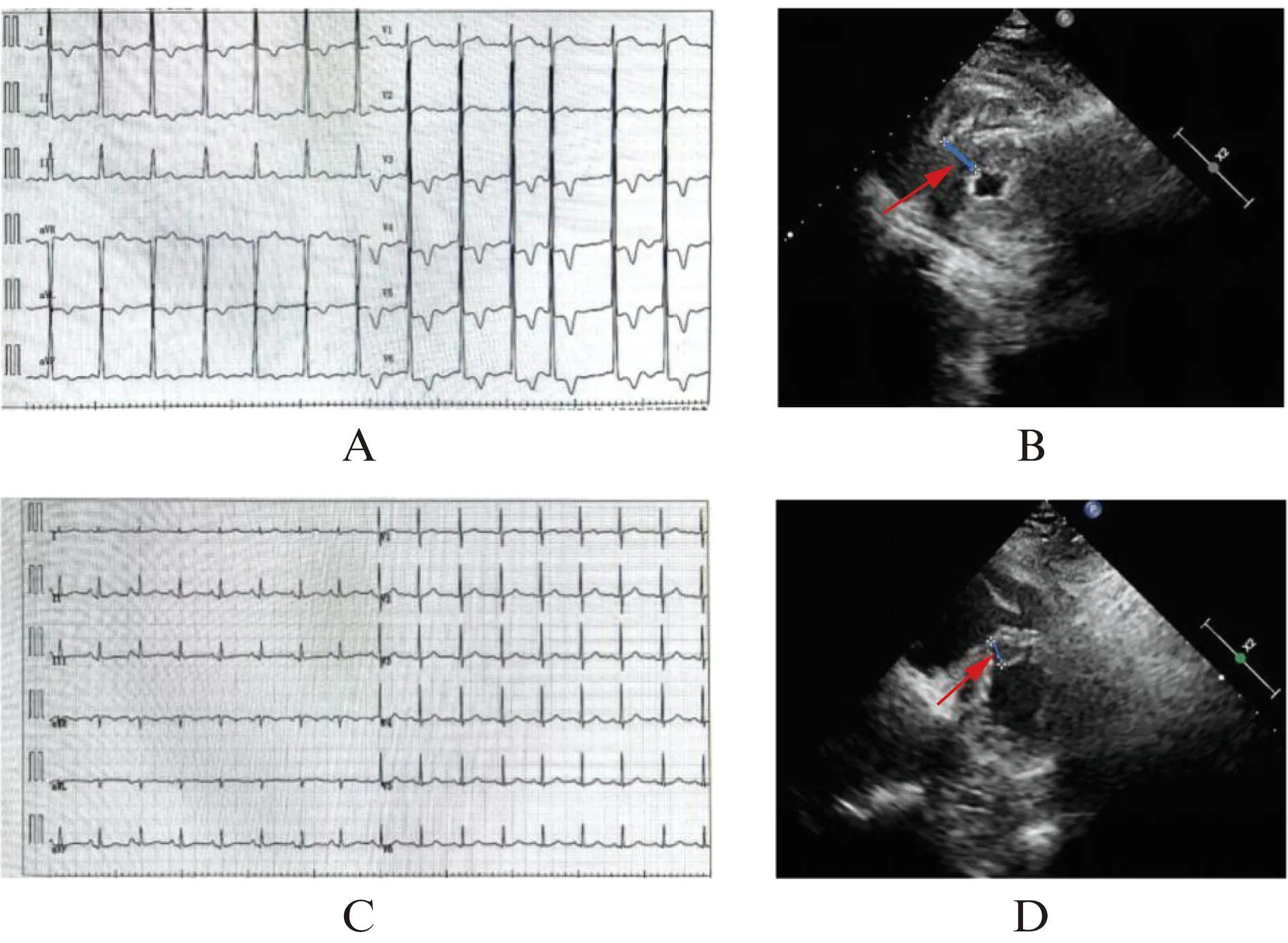
图4 先证者及家系成员的影像学图像
在家系筛查中,发现先证者长子(Ⅲ-1)41岁出现活动后胸闷,46岁时心电图显示窦性心律、左心室肥大、ST-T改变,超声心动图显示室间隔厚度为17.0 mm,左室后壁厚度为11.0 mm,左心室射血分数64.0%;次子(Ⅲ-3)43岁时开始出现活动后气促,48岁时心电图显示窦性心律、左心室肥大;超声心动图显示室间隔为14.4 mm,左室后壁厚度为10.8 mm,左心室射血分数58.1%,见表1。两者均表现为不对称性室间隔肥厚,同时未发现导致心肌肥厚的继发性因素。进一步行Ⅲ-1和Ⅲ-3的测序验证,发现两位均携带与先证者相同的MYBPC3-D1149fs*40突变。对该家系患者进行动态心电图监测未发现恶性心律失常,家族中无猝死病例。
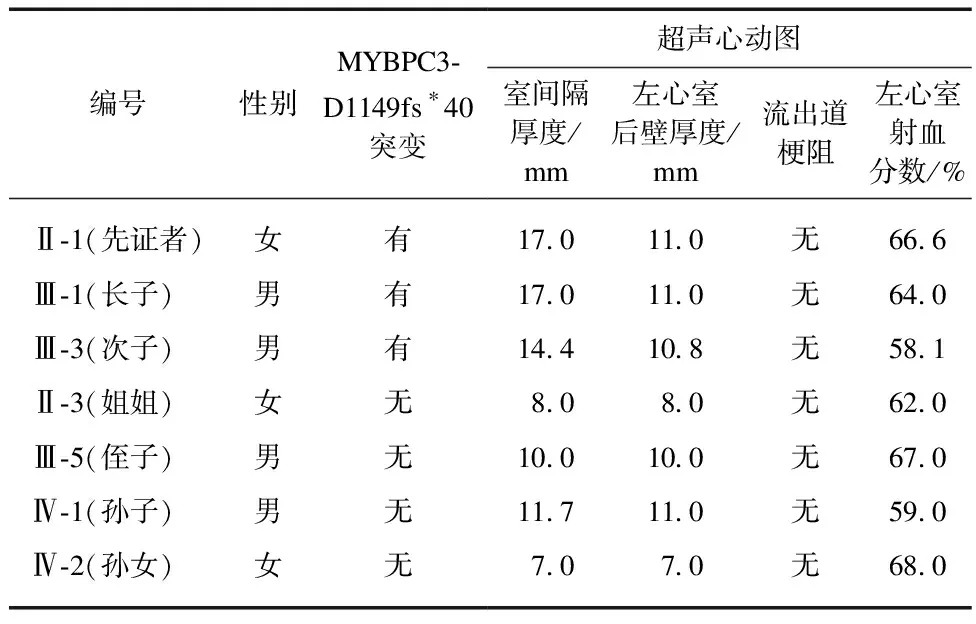
表1 家系成员的超声心动图特点
筛查的其余家系成员(Ⅱ-3、Ⅲ-5、Ⅳ-1、Ⅳ-2)无不适主诉,超声心动图和心电图正常,见图4C、D;同时测序未发现MYBPC3-D1149fs*40突变。
3 讨 论

本中心针对2023年2月就诊于我院心内科的1例HCM患者及其家庭成员进行临床资料采集和基因学分析。先证者为老年女性非梗阻性肥厚型心肌病患者,检测到MYBPC3的c.3445(exon 31)delG(p.Asp1149fs*40)基因突变,即第31号外显子上的3 445位碱基G缺失,导致第1 149位天冬氨酸的编码序列出现移码突变,在移码突变后方生成40个氨基酸后提前引入终止密码子,从而终止转录翻译过程,导致肽链缩短,无法完整表达蛋白。基因检测发现该患者的长子和次子均携带相同的MYBPC3-D1149fs*40突变。携带该突变的3位家系成员均表现为不对称性室间隔肥厚,平均室间隔厚度(16.1+1.2) mm,室间隔与左室后壁厚底之比均大于1.3,病史询问、体格检查及相关辅助检查未发现高血压、主动脉瓣狭窄等导致心肌肥厚的因素,符合HCM的诊断标准[1]。3位患者的平均发病年龄为(49.3±10.4)岁,均不伴有左室流出道梗阻,病程中无黑矇、晕厥,动态心电图监测未发现恶性心律失常,家族中无猝死病例。这符合MYBPC3基因突变携带者发病时间较晚(>40岁)、症状轻、晕厥及猝死等并发症发生率低的特点[6-7]。
在该家系的另4位临床表型完全正常的成员中未检测到MYBPC3-D1149fs*40基因突变,结合该家系3例HCM患者携带该突变位点,即突变与HCM临床表型存在共分离现象,提示MYBPC3-D1149fs*40突变为该家系患者的致病基因突变。该突变位点在dbSNP、千人基因组等均未被收录,考虑为MYBPC3基因新的突变位点。氨基酸保守性分析表明,该突变在许多物种中高度保守。使用SIFT、Polyphen2等多软件对该位点进行致病性预测表明,该位点的突变为致病性突变。
本研究中发现的MYBPC3基因G碱基缺失导致读码框破坏,产生读码框的移码突变,使转录翻译提前终止,导致由MYBPC3基因编码的心脏型肌球蛋白结合蛋白C(cMYBP-C)无法完整表达,cMYBP-C的羧基末端(C端)异常缺失[2,4-9],缺乏主要的肌球蛋白和肌动蛋白结合位点,不能发挥固定粗肌丝的作用,造成肌小节结构和功能异常,因此可能引起HCM[10]。
综上所述,本研究在一家族性HCM患者中发现MYBPC3-D1149fs*40的新发变异,且该突变在该家系中呈共分离现象,推测MYBPC3-D1149fs*40突变是该HCM家系的致病突变。
