石墨炉原子吸收法测定水中铍的不确定度评定
2024-01-10梁坤玲
梁坤玲
(广东省科学院测试分析研究所(中国广州分析测试中心),广东 广州 510000)
传统的误差评定,总是会在实践检测过程中遇到不同实验员对误差不同的处理方式,从而使实验员测量出来的结果缺少可比性及概念上不一致这两个问题,为了解决误差评定的困难与局限,能更好统一评价的测量结果,从而产生了测量不确定度。实现了可在各个领域采用的用于表示和评估测量结果的实施标准。《测量不确定度指南》[1]提供了测量不确定的表示方法及评定。测量不确定度从本质上改变了将测量误差分为随机误差和系统误差的传统分类方法,是经典误差理论发展和完善的产物。由于不确定度作为未确定误差的特殊表示,因此其不能用于对测量结果的修正。测量不确定度可在可修正的系统误差后进行修正[2]。因为测量不确定度比经典的误差表示方法更为科学使用,所以在世界各国都广泛使用。
不确定度是一个与测量结果有直接关系的参数,用来表示被测量者分散性的相关数值[3]。从词义上理解测量不确定度,就是对测量出来的结果由于测量误差的存在而不能肯定的程度,反过来,也可表征该测量结果的可信程度,是一个用数字表示测量结果质量的参数。在实际检测中由于人们的认知不全及测量方法的不完善,所导致被测量值具有不唯一性,多次测量下,多个数值均匀分布在某个区域内。尽管系统误差作为客观存在的一个常数,但我们不能完全认知或把握它,只能认为系统误差是一定条件下随机地分布在某个区域内。
由于无法通过实验测量获得测量是真实值,我们只能借助测量不确定度来知道被测溶液的测量值在什么范围内,测量不确定还可以说明一个实验室的分析水平程度。
铍是一种具有特殊性质的化学元素,由于它具有许多优异的物理和化学性能,如核性能和物理性能,从而在原子能、激光器件、家用电器、导弹、航空、火箭、宇宙行业以及治金工业中是其他金属元素不可取代的。然而,铍是在一类致癌物清单中剧毒的金属元素之一。铍化合物主要以呼吸道、创伤表皮及口腔进入人体,在人体内沉积会对细胞组织造成广泛损伤,尤其对皮肤和肺部的危害最为显著,其病症包括眼结膜炎、皮肤溃烂、支气管炎、呼吸系统的鼻黏膜炎、支气管喘息、接触性皮炎、肺气肿、活动性肺结核、咽炎、化学性肺炎,肺部的长期延续性病变[4-5],发病时间可延缓至接触铍化合物后的20年。铍工业的发展也一直受铍毒性的影响。随着人类对环境保护的越发严格,进而提高对铍毒性的防护。目前铍毒性的防护主要采用加强通风与粉尘收集来防止毒物的扩散,可惜铍毒性的防护问题至今尚未彻底解决,仍然是阻碍铍工业发展的罪魁祸首。在正常情况下,水中的铍元素的含量相对较低,为了使水中铍元素更为准确地测量,测量水中铍元素的不确定度就显得尤为重要。
《测定水中铍元素含量不确定度的方法:石墨炉原子吸收法》以实例介绍了石墨炉原子吸收分光测定法,其合理评定不确定度的大小决定了检测结果的准确性。
1 检测方法
1.1 检测方法依据
依据行业标准《水质 铍的测定 石墨炉原子吸收分光光度法》(HJ/T 59—2000)[6],科学评估水中铍元素的不确定度。
1.2 检测方法原理
先将原子化放在热解石墨炉中进行加热处理,吸收由空心阴极灯发射的特征辐射,它就可以成为基态原子蒸气。在某个特殊范围中,溶液的吸收强度越大则该试液铍元素的含量就越高[7]。
1.3 实验主要使用的仪器
HITACHI(日立)Z - 2010型原子吸收分光光度计。
1.4 实验步骤
1.4.1 铍标准曲线的绘制
1.4.1.1 铍标准使用液的配制
在实验过程中,实验员用1 000 mg/L的铍标准贮备液,根据行业标准HJ/T 59—2 000进行稀释。将铍标准贮备液,依次逐级稀释至0.10 mg/L。
1.4.1.2 铍标准曲线的绘制
准备一组 7个100.00 mL洁净的容量瓶,依据行业标准HJ/T 59—2000,依次加入铍的标准使用液 0.00,0.50,1.00,1.50,2.00,2.50,3.00 mL,然后加入0.5 mL的0.10 mg/L硝酸铝溶液和2.0 mL的(1+1)硫酸溶液,加水至标线,定容,得到多个不同浓度的标准溶液,如0.0,0.50,1.00,1.50,2.00,2.50,3.00 mg/L等标准溶液,由低浓度溶液依次向高浓度溶液测定其的吸光度,编制健全的标准曲线。
1.4.1.3 样品测定
将50 mL水样移加到150 mL的锥形瓶中,移取5 mL的浓硝酸溶液加入锥形瓶中,移至电热板上控制温度,在锥形瓶表面覆盖小漏斗,将溶液温度长期控制在(95±5) ℃,使溶液一直处在不沸腾的状态,设置电热板加热时间,使回流时间控制在30 min,将小漏斗拿开,持续加热,待溶液蒸发至大约5 mL,停止加热。等到溶液完全待冷却后,再移取5 mL浓硝酸溶液加入其中,用小漏斗盖住,回流继续加热,如此反复,直至棕烟不再产生。待冷却后,3 mL过氧化氢溶液加入其溶液中,覆盖小漏斗,使溶液温度控制在95 ℃左右,直至不冒大量气泡为止。在其完全溶液冷却后移除小漏斗,继续加热至溶液大概在5 mL,溶液冷却后,至少用水冲洗内壁3次,然后加入0.5 mL的0.10 mg/L硝酸铝溶液和0.2 mL的(1+1)硫酸溶液,定容至50 mL容量瓶中。
2 数学模型
校准曲线拟合的回归方程[8]:
y=a+bx
式中:y—-待测溶液的吸光度;
x—待测溶液铍元素的浓度值,mg/L;
a—回归拟合线方程的截距;
b——回归拟合线方程的斜率。
水中铍元素浓度的计算公式为:
C=x/Va×Vd
式中:Vd—溶液的定容体积,mL;
Va—测定水样的体积,mL;
x—样品铍元素的浓度值,mg/L;
C—水样中铍元素的浓度,mg/L。
根据上述公式计算可知,配置的标准溶液、取样体积、定容体积及工作曲线的拟合样品的重复测量可影响测量结果的不确定度,且每一个不确定度分量都是互不相连的。基于不确定传播规律的基础上,建立不确定度数学表达式:
式中:urel(c)表示水样中铍元素浓度的不确定度;
urel(f)表示样品重复测定的不确定度;
urel(V)表示铍标准溶液的不确定度;
urel(m)表示绘制标准曲线的不确定度;
urel(A)表示计算体积的不确定度;
urel(R)表示计算水样定容体积的不确定度;
urel(S)表示计算样品前处理消解过程中的不确定度。
3 分析不确定度分量主要来源
不确定度的来源有以下几个方面:[9]1)在使用配制铍标准贮备液和稀释铍标准贮备液至铍标准使用液过程中所产生的不确定度;2)样品取样体积中所产生的不确定度;3)样品前处理解消过程中所产生的不确定度;4)样品重复测定所产生的不确定度,由样品溶液的重复性进行多次测定而得;5)溶液定容体积中所产生的不确定度。
4 评定不确定度分量
4.1 铍标准贮备液所产生的不确定度
实验员在通过实验分析铍标准贮备液时发现其浓度为
1 000 mg/L的标准溶液,不确定度仅为4 mg/L。实验员根据正态分布进行计算,得出:
urel(V1)=u(V1)/ccr/k=4/1 000/2=0.002
4.2 标准贮备液稀释不确定度
4.2.1 玻璃器皿中液体的不确定度
4.2.1.1 单标线移液管中液体的不确定度

4.2.1.2 100 mL容量瓶中的不确定度
100 mL容量瓶中不确定度计算如表1。

表1 玻璃器皿产生的不确定度
通过分析上表内容,得到容量瓶不确定度:
4.2.2 将1 000 mg/L铍标准贮备液稀释至0.10 mg/L铍标准使用液四次稀释的过程中所产生的不确定度
1)1 000 mg/L铍标准贮备液稀释至100 mg/L铍标准中间使用液的不确定度:根据行业标准HJ/T 59 — 2000进行稀释,用10.00 mL单刻度移液管,移取10.00 mL的1 000 mg/L铍标准贮备液到100 mL单标线容量瓶中,用水稀释至刻度线,定容。
2)100 mg/L铍标准中间使用液稀释至10 mg/L铍标准中间使用液产生的不确定度:
根据行业标准HJ/T 59 —2000进行稀释,稀释过程如上,即:
urel(V5)=urel(V4)=0.001 0
3)10 mg/L铍标准中间使用液稀释至1.0 mg/L铍标准中间使用液产生的不确定度:
根据行业标准HJ/T 59 — 2000进行稀释,稀释过程如上,即:
urel(V6)=urel(V4)=0.001 0
4)1.0 mg/L铍标准中间使用液稀释至0.10 mg/L铍标准使用液产生的不确定度:
根据行业标准HJ/T 59 — 2000进行稀释,稀释过程如上,即:
urel(V7)=urel(V4)=0.001 0
铍标准溶液配制的不确定度为:

4.3 标准曲线拟合所产生的不确定度
依据行业标准进行稀释配制而成铍的标准系列由仪器进行自动测量,测量结果见表2。

表2 铍标准曲线测量结果
仪器自动得出:a=0.022 9,b=0.146,r=0.999 5。得到回归方程:
y=0.146x+0.022 9
根据贝塞尔公式,将上述相关数据代入其中,计算剩余的标准偏差:
单次测量结果标准曲线拟合产生的标准不确定度:
式中:p—测定样品的次数,次数为7;
x—多次测量的水样中铍浓度的平均值,x=0.90 μg/L;

b—曲线斜率,b=0.146;
n—测定标准溶液的次数,次数为7;
xi—溶液浓度值,mg/L。
则计算出水样中铍的相对标准不确定度为:
4.4 样品重复测量所产生的不确定度
对该水样进行重复7次的测量,测量结果见表3。

表3 水样中铍元素含量测定结果
按公式计算出样品重复测量的不确定度为:
4.5 样品消解过程中所产生的不确定度
在水样消解过程中,很容易受到各种外在因素所影响,如样品复杂程度、消解条件等因素,从而出现大量不完全消解的现象,无形中增加样品消解过程中铍含量的随机性,实验员利用加标回收率实验评估其不确定度;根据HJ/T 59—2000石墨炉原子吸收分光度法测定水的铍含量,加标回收率范围在94%~113%,已知加标回收率范围上、下界相对于最优值呈非对称分布,计算出加标回收率的不确定度如下:
铍加标回收率要求控制在94%~113%范围内,理论上的加标回收率最佳值为100%,因此,b+为113%-100%=13%,b-为100%-94%=6%,则样品消解过程中所产生的不确定度:
4.6 取样体积所产生的不确定度
4.7 定容体积所产生的不确定度
50 mL容量不确定度如表4。

表4 50 mL容量瓶产生的不确定度
urel(R)=urel(A)=0.000 46
5 合成不确定度
合成相对标准不确定度为:
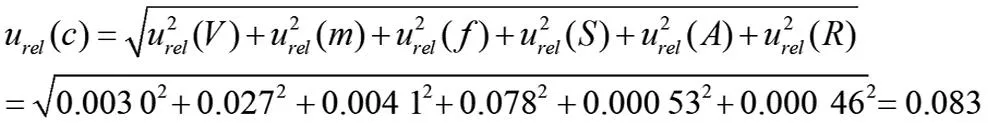
则合成标准不确定度为:
6 扩展不确定度
因子k=2(约95%置信概率),则扩展不确定度为[10]:
U=kuc(c)=2.0×0.075 μg/L=0.15 μg/L
7 结论
利用行业标准石墨炉原子吸收分光光度法测定水中铍的不确定度,测定结果为0.90 μg/L,扩展不确定度数为0.15 μg/L(k=2),或以测量结果表示为(0.90+0.15)μg/L,k=2。
通过对不确定度的计算及分析,最终确定主要影响测量结果的是标准曲线拟合及样品前处理消解过程。针对该种情况,实验员应该注重通过提高方法准确度、灵敏度、减少样品前处理消解的过程的损失等方法,全面增加数据的可靠性。
