1-甲基海因对慢性疼痛大鼠钠离子通道蛋白Nav1.8的干预作用
2024-01-10梁雾滢曾玉淇吕俊瑾莫睿文远立国
梁雾滢,刘 镇,曾玉淇,吕俊瑾,莫睿文,远立国*
(1.华南农业大学兽医学院,广州 510642;2.广东省兽医临床重大疾病综合防控重点实验室,广州 510642;3.广东省宠物工程技术研究中心,广州 510642)
慢性疼痛为持续或者反复发作时间超过3个月,与潜在或实际组织损伤有关的不良情感体验。因发生率高,病因复杂,机制尚未明晰,目前尚无科学有效的干预措施,慢性疼痛已成为威胁健康的关键因素之一[1-3]。科学干预是使“疼痛最小化”唯一途径[4],药物干预是兽医临床慢性疼痛治疗的主要方法,虽可抑制疼痛反应,但尚存在不同程度的不良反应。
慢性疼痛过程涉及多种介质[5],Nav1.8作为电压门控钠离子通道(voltage-gated sodium channels, VGSC)的一种亚型,在慢性疼痛过程参与度较高,优先分布于外周感觉系统的小直径神经元中,参与钠离子内流过程,在外周神经敏化中作为重要的离子通道发挥作用[6-7]。VGSC阻滞剂镇痛效果明显[8],但广谱钠通道阻滞剂并非仅阻滞与疼痛相关的亚型,同时会阻滞与骨骼肌、心肌相关的亚型并产生严重的副作用。阻滞Nav1.8后不会影响机体主要的生理功能,且Nav1.8结构和功能的改变在慢性疼痛的发展中发挥关键作用,因此已成为治疗慢性疼痛备受关注的靶点[9-10]。
海因类化合物作为钠离子通道阻滞剂或一种非肽类生长抑制素,受体配体可高度选择性作用于SSTR2,具有良好的镇痛作用[11],通过结构改造或修饰可以克服现有不足[12],有望成为治疗慢性疼痛的先导化合物[13]。MH作为哺乳动物的代谢产物,属海因类最简单的小分子化合物,价廉、易得,若MH可通过选择性阻滞Nav1.8的表达而起到镇痛效果,其成药潜力巨大。因此本文通过MH对坐骨神经慢性压迫模型(chronic constriction injury, CCI)大鼠慢性疼痛的作用,探究MH镇痛的可能性。
1 材料与方法
1.1 材料
Nav1.8通道的CHO细胞系(中国SCOPE公司);1-甲基海因(CATO);氨溴索(上海甄准生物科技有限公司);二甲基亚砜(北京索莱宝科技有限公司);水合氯醛(天津大茂化学试剂厂);兔多克隆抗体to Nav1.8、山羊抗兔IgG抗体(英国Abcam公司)。
1.2 方法
1.2.1 实验动物分组及建模 SPF级SD大鼠60只,体重(250±10)g,广东省医学实验动物中心提供[生产许可证号:SCXK(粤)2016-0041],饲养于华南农业大学实验动物中心。伦理批文号:2018B092。大鼠按雌、雄各半随机分为正常对照组(N组)、假手术组(P组)、模型对照组(M组)、1-甲基海因组(MH组)、氨溴索组(A组)、利多卡因组(L组),每组10只。参照Bennett 等[14]的方法建立CCI模型;假手术组暴露坐骨神经但不结扎;正常对照组不作任何处理。MH组、A组和L组大鼠在建模后第10天开始,直至第21或28天取DRG为止,分别接受肌注MH(25 mg·kg-1)、口服氨溴索(1 000 mg·kg-1)以及肌注2%利多卡因(0.2 mg·kg-1)治疗。
1.2.2 蛋白免疫印迹 经NCBI查询获得Nav1.8的蛋白序列,根据序列通过Expasy计算出通道分子量。从分离出的大鼠背根神经节中提取总蛋白并测定蛋白浓度,蛋白样品经电泳后转至聚偏二氟乙烯膜于4 ℃封闭2 h ,用TBST缓冲液1∶4 000配制兔多克隆抗体to Nav1.8,4 ℃孵育12 h,山羊抗兔IgG抗体按1∶5 000稀释,4 ℃避光孵育6 h,洗膜后利用红外荧光扫面成像仪器进行扫膜,以β-actin为内参,分析相关蛋白的表达水平。
1.2.3 全细胞膜片钳技术 根据全细胞膜片钳技术检测经MH孵育的CHO细胞电生理变化,选用利多卡因作为阳性对照化合物,检测其对Nav1.8电流的抑制率。

2 结 果
2.1 蛋白免疫印迹试验
建模后第21和28天,与P组相比,手术各组大鼠DRG中Nav1.8的表达均提高,未用药的M组Nav1.8表达最多。用药各组DRG中Nav1.8表达量依次为MH组、L组、A组。用药各组与M组比较,Nav1.8表达量均显著降低(图1和图2,P<0.05)。N组与P组比,差异不显著(图1和2,P>0.05)。
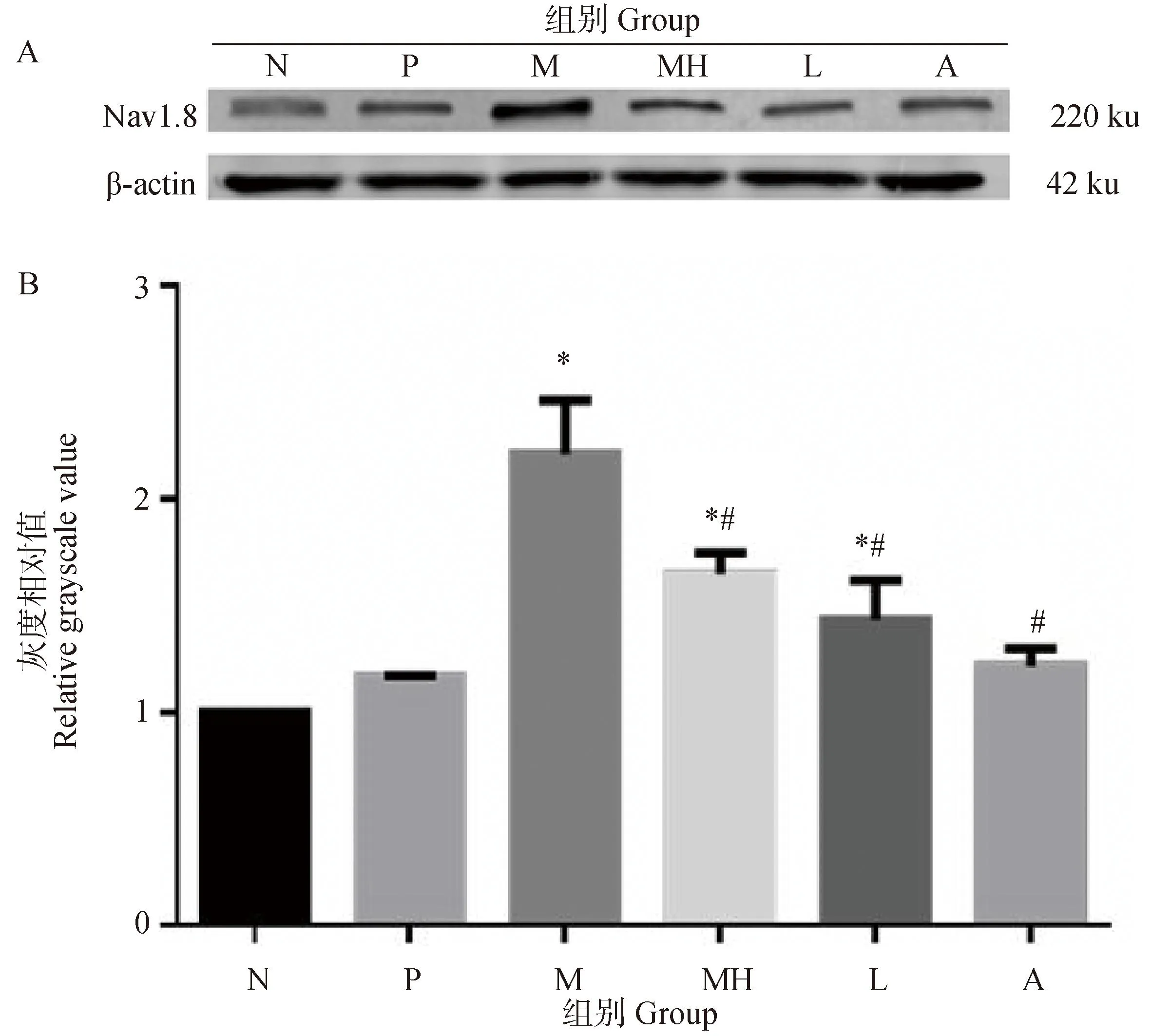
A. Nav1.8表达量;B.灰度相对值分组介绍: N. 正常对照组; P. 假手术组; M. 模型对照组; MH. 1-甲基海因组; A. 氨溴索组; L. 利多卡因组,各组均与N组比较,*. P<0.05;用药组与M组比较,#. P<0.05A. Expression of Nav1.8; B. Relative grayscale Group introduction: N. Normal control group; P. Sham surgery group; M. Model control group; MH. 1-methylhyne group; A. Ambroxol group; L. Lidocaine group, all groups were compared with group N, *. P<0.05; The treatment groups compared with group M, #. P<0.05图1 第21天大鼠DRG中Nav1.8的表达量及灰度相对值Fig.1 The expression of Nav1.8 and relative brightness DRG of rats on the 21st day

A. Nav1.8表达量;B.灰度相对值分组介绍: N. 正常对照组; P. 假手术组; M. 模型对照组; MH. 1-甲基海因组; A. 氨溴索组; L. 利多卡因组,各组均与N组比较,*. P<0.05;用药组与M组比较,#. P<0.05A.Expression of Nav1.8; B.Relative brightness Group introduction: N. Normal control group; P. Sham surgery group; M. Model control group; MH. 1-methylhyne group; A. Ambroxol group; L. Lidocaine group, all groups were compared with group N, *. P<0.05; The treatment groups compared with group M, #. P<0.05图2 第28天大鼠DRG中Nav1.8的表达量及灰度相对值Fig.2 The expression of Nav1.8 and relative brightness DRG of rats on the 28th day
2.2 全细胞膜片钳技术检测
10和100 μmol·L-1MH对Nav1.8的电流抑制率分别为6.34%±3.13%和14.6%±1.52%,阳性对照化合物100 μmol·L-1利多卡因对Nav1.8的抑制率为57.2%±2.23%(图3和4),因此抑制Nav1.8通道电流抑制强度依次为100 μmol·L-1利多卡因、100 μmol·L-1MH、10 μmol·L-1MH。
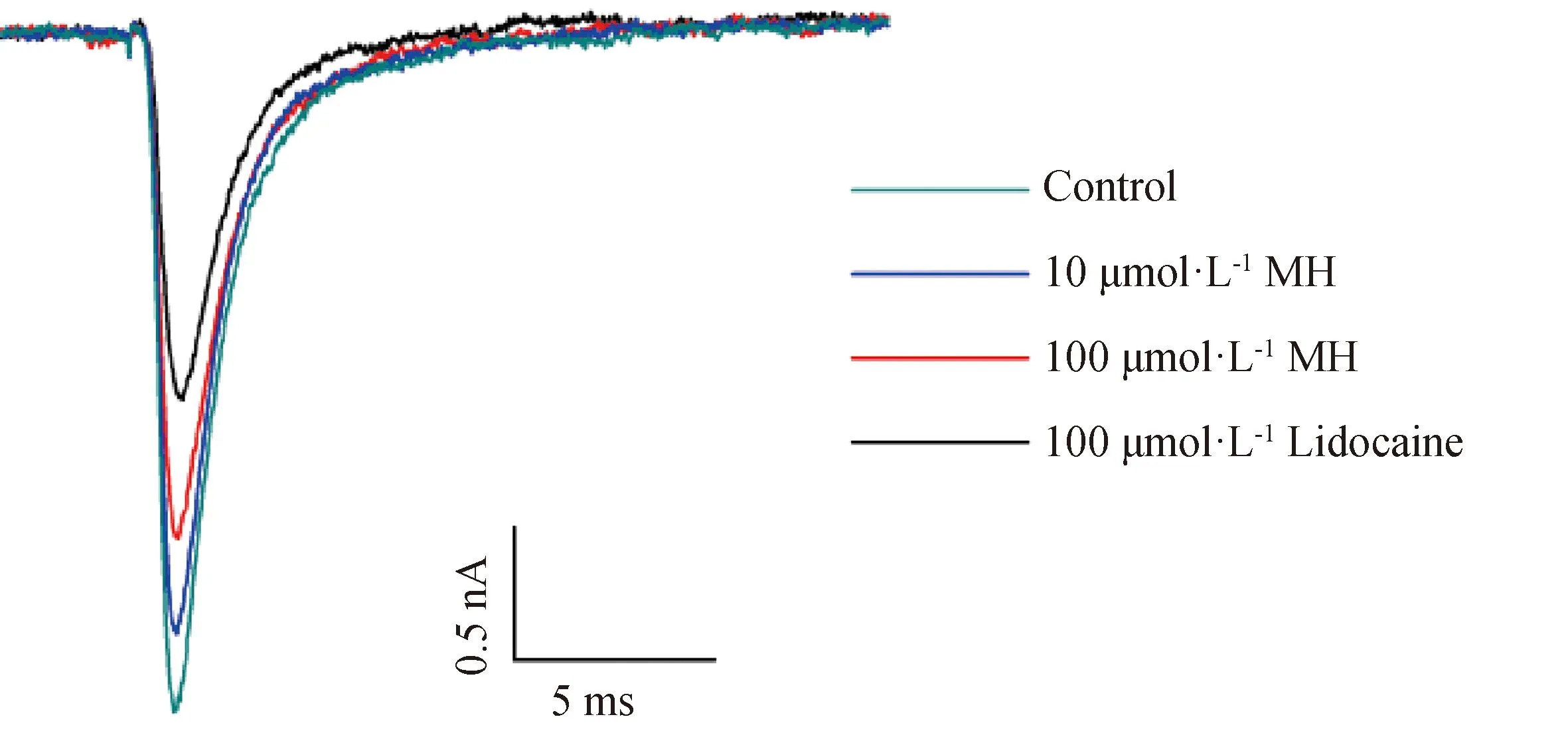
Control为正常外液记录的电流,然后依次灌流10、100 μmol·L-1 MH,洗脱后给予100 μmol·L-1利多卡因Control was the current recorded from the normal external fluid, and then 10 and 100 μmol·L-1 MH were successively perfused, and 100 μmol·L-1 lidocaine was administered after elution图3 抑制Nav1.8通道电流图Fig.3 Pattern of inhibition of current of Nav1.8 channel
3 讨 论
本试验通过MH和Nav1.8调节剂干预大鼠慢性疼痛后脊髓背根神经节的变化,使用蛋白免疫印迹技术检测大鼠L4~L6 段DRG中Nav1.8的蛋白表达,采用全细胞膜片钳技术检测MH作用下的Nav1.8通道峰电流,初步探究MH对慢性疼痛大鼠Nav1.8的作用。
疼痛造成了动物DRG中Nav1.8蛋白表达量上调,可能是因为慢性疼痛造成DRG中传入感觉神经元上的Nav1.8表达增加,但随着时间推移,机体自身免疫系统参与抗炎,炎症减轻,蛋白表达下降[15-17]。
钠离子通道结构和功能的改变影响疼痛转归过程,利多卡因通过调节钠离子通道状态而产生镇痛效果,MH和利多卡因均抑制了Nav1.8的活性,表明这两种化合物的镇痛机制可能涉及钠离子通道,也说明抑制Nav1.8可能与镇痛反应有关,但也可能涉及多种复杂机制。本试验测得氨溴索抑制Nav1.8蛋白表达的效果强于利多卡因,但镇痛效果则是利多卡因最佳,其原因可能是氨溴索是Nav1.8高选择性阻滞剂,而造成疼痛的机制复杂,抑制Nav1.8的表达只是其中的一个通道,利多卡因还可拮抗其它通道如Nav1.9的表达而起到更好的镇痛效果[18]。
神经或组织损伤后,Nav1.8直接参与DRG神经元异常电活动,产生异常的钠离子电流,这可能是因为吸引交感神经纤维包裹成篮状结构,继续产生异常的细胞膜电位[19]。MH对Nav1.8通道电流具有抑制作用,且呈现浓度依赖性。将暴露于100 μmol·L-1MH的CHO细胞洗脱,该细胞的Nav1.8通道几乎可以从失活状态恢复到静息状态,说明100 μmol·L-1MH对Nav1.8的抑制作用基本可逆。相较于100 μmol·L-1酚那酸类药物作用于CHO细胞洗脱后,CHO细胞的Nav1.8通道难以从失活状态恢复到静息状态,抑制作用不完全可逆[20]。证明了在用药后MH对Nav1.8通道电流具有抑制作用,且基本不影响疼痛恢复期Nav1.8的功能,说明MH在治疗慢性疼痛过程中的安全性。
目前,已有Nav1.8阻滞剂进入临床试验,但数量十分有限。本研究证明,当浓度从10 μmol·L-1升至100 μmol·L-1,MH对Nav1.8抑制率仅从6.34%±3.13%增长到14.6%±1.52%,表明MH对 Nav1.8抑制效果较弱,但由于MH结构简单,廉价易得,通过结构改造或修饰可以克服现有不足,因此仍然具有成为动物镇痛药物的潜力。目前,已确定磺胺类镇痛药与钠离子通道蛋白的结合位点,并且与经典的小分子钠通道阻滞药如利多卡因等物不同,后者作用于钠离子通道的孔隙区域,所以通常对不同的钠通道亚型选择性很小,故后续可能通过利用磺胺类化合物的高特异性结合位点,对海因类化合物类镇痛药进行筛选设计和改造。
本试验对1-甲基海因治疗大鼠CCI模型致慢性疼痛的进行了初步研究,发现MH可以抑制Nav1.8蛋白表达,同时抑制钠离子电流。但本研究具有一定的局限性,Nav通道各亚型之间具有高度相似性[21],后续可检测模型动物DRG中Nav1.8基因的表达情况,分析MH对其余钠通道亚型的阻滞作用和选择性,为后期基于MH结构进行改造或修饰,从而进一步研发出特异性高、副作用少的新型镇痛药提供依据。
4 结 论
MH通过下调慢性疼痛大鼠DRG中Nav1.8的表达,同时抑制Nav1.8通道电流,进而产生的镇痛作用,故MH具有成为动物镇痛药物的潜力,本试验为临床科学治疗慢性疼痛和基于MH进行动物专用新型镇痛药物的研发提供理论依据。
