有机液体储氢体系研究进展
2024-01-07李欣雨唐鋆磊李佳奇雷宪章周太刚
李欣雨,唐鋆磊,2,李佳奇,雷宪章,2,4,周太刚,2,4
(1.西南石油大学 化学化工学院,碳中和研究院,四川 成都 610500;2.天府永兴实验室,四川 成都 610000;3.中国农业大学 理学院,北京 100193;4.成都岷山绿氢能源有限公司,四川 成都 610000)
随着社会的不断发展,人类对化石能源的需求不断增长,导致温室气体排放量逐渐增多。联合国政府间气候变化专门委员会(ⅠPCC)第6 次评估报告指出,近年来人为温室气体排放量达到了历史最高值,气候变化已经对陆地、淡水、沿海和远洋海洋生态系统造成了巨大破坏和越来越不可逆转的损失,温室效应引发的生态环境恶化问题日益严峻。为应对全球气侯变化,实施有效可行的温室气体减排措施迫在眉睫,为此中国政府承诺,力争2030 年达到碳达峰,2060年实现碳中和[1-3]。要实现双碳目标,能源变革是重中之重。然而,能源结构由化石能源向清洁能源的转换,将使电力系统面临极大的挑战:在大规模应用新能源的条件下,传统燃煤电站将成为辅助电源以及大规模外送电力的馈入,电力系统将面临“空心化”,而高比例的风光能源的间歇性和波动性,使电力系统的功率稳定性具有了较大的不确定性,对系统的动态稳定运行也形成了巨大挑战。此外,在重大自然灾害情况下如何应对电网的供电可靠性,也是在能源转换过程中需要解决的重大问题[4]。因此,迫切需要发展与能源结构转换相适应的新型储能技术。氢能是真正意义上清洁环保的二次能源,具有来源充足、清洁低碳、高热值和高转化率等多方面优势。将氢能作为能源载体,桥接传统能源和其他低碳能源,是在减少对传统能源依赖的同时解决低碳能源区域限制、季节限制问题的重要方法之一,发展氢电耦合是保障新型电力系统稳定运行的必要途径。然而,氢气较宽的爆炸极限范围(体积分数为4.0%~75.6%,遇火源就会爆炸)以及极低密度(在0 ℃、101 kPa的条件下,氢气密度为0.0899 g/L)使氢能产业链发展至今,呈现出制氢易、储氢难和用氢更难的特点。其中,氢气储运的技术挑战和经济压力是制约氢能产业大规模发展的关键因素。目前,按照不同的储氢机理,可以将储氢技术分为高压气态储氢、低温液化储氢、固体材料储氢以及有机液体储氢(LOHC),不同储氢技术的对比见表1。
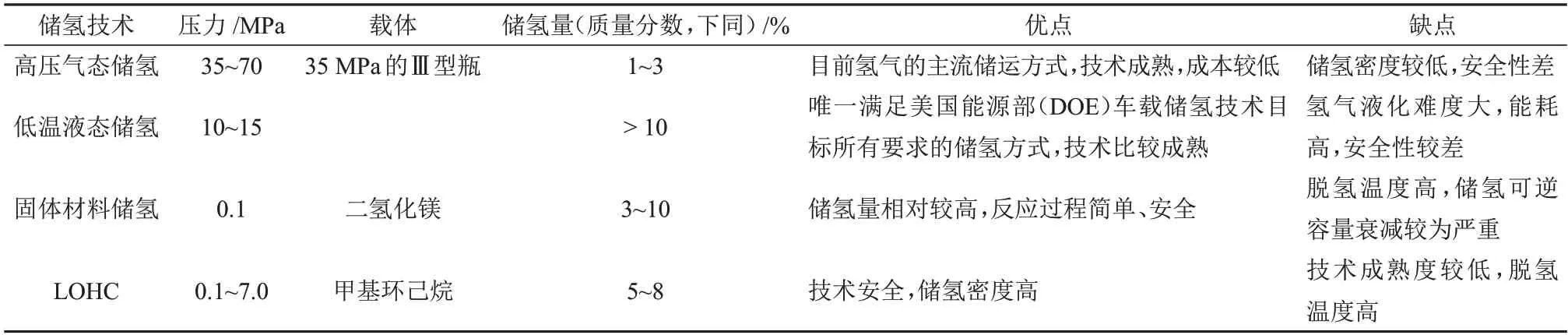
表1 不同储氢技术的对比Table 1 Comparision of different hydrogen storage technologies
从表1 可以看出,高压气态储氢是目前应用最广的储氢技术,技术成熟,储放氢操作简单。但该储氢技术的氢气密度受压力和储氢气罐两个因素影响。储存的氢气密度与压力成正比,压力越大,氢气密度越高,当压力在30~40 MPa时,氢气密度增加较快,当压力大于70 MPa时,氢气密度变化较小。高压气态储氢对储氢气罐材质要求严苛,在不同压力条件下,需要使用与压力相匹配的耐压容器。目前用于工业或运输的氢气通常需压缩到15~20 MPa的压力,但是1个充气压力为20 MPa的标准高压钢瓶储氢量仅约1.0%,因此高压气态储氢的氢气能量密度一般都比较低。在低温液态储氢技术中[5],为使氢气液化首先需要将温度降低到-253 °C以下,然后实施多次的绝热膨胀。液氢的沸点(-252.78 °C)极低且容易汽化,为了避免液氢因汽化造成的损失,储存液氢的容器大多采用多层绝热材料制造。氢气的液化和液氢的储存过程需要耗费大量的能量,生产和储存的经济成本高,但是低温液态储氢技术的氢气能量密度是所有储氢技术中最高的,目前只在航空航天领域有完整应用体系。固体材料储氢技术[6]无需高压及绝热容器,具有氢气密度高、运行压力低和安全性好等特点,但是固体材料在脱氢反应过程中,常伴随着副反应发生,储氢可逆容量衰减严重。固体材料储氢技术目前尚处于实验研究阶段,主要的潜在应用领域为交通、储能和应急发电等。
1975 年,SULTAN 等[7]基于Π-共轭体系中共轭效应使体系氢化热降低的特点,借助有机液体化合物(环烷烃催化加(脱)氢)开展了汽车储氢研究,首次提出了LOHC概念,为氢气储运提供了一种新方法。LOHC技术的原理见图1,烯烃、芳烃等有机化合物与氢气发生催化加氢反应,生成相应的储氢载体,储氢载体通过催化脱氢反应,将氢气释放出来,而LOHC载体被回收。通过这种方式,储氢材料可以多次使用而不被消耗,储氢量大、能耗相对较低且安全性较好[8-9]。继SULTAN 等提出LOHC 概念后,2002年日本千代田化工建设公司进行了基于甲苯的LOHC系统开发,针对甲苯-甲基环己烷储氢体系低温转化率低和脱氢催化剂易失活等问题研发相对应的高效催化剂。2009 年WANG 等[10]报道了首例催化剂作用下的N-乙基咔唑(NEC)可逆加(脱)氢反应,发现铱配合物催化剂对全氢化N-乙基咔唑(12H-NEC)脱氢具有较高催化活性,然而同时也发现12H-NEC 因受到高脱氢焓热力学约束很难生成完全不饱和的NEC。同年日本千代田化工建设公司在LOHC系统的关键技术方面取得突破,发展了新型Pd 基脱氢催化剂,在示范运行时,将脱氢催化剂的“有效寿命”延长至1年以上。2013年德国Hydrogenious Technologies 公司基于二苄基甲苯良好的热稳定性和独特的理化性质,开发了二苄基甲苯LOHC 储氢体系,借助高活性催化剂优化二苄基甲苯储氢材料的生命周期和效率。2014 年武汉氢阳能源控股有限公司基于含氮杂环低温脱氢的优势,开发了NEC储氢体系。近年来LOHC技术发展迅速,取得了一系列研究成果,示范进程也在推进。2022年,日本千代田化工建设公司利用甲基环己烷储氢体系,将甲基环己烷通过化学品运输船运往海外,实现了LOHC 技术海上运输全球首个示范,证明了该技术的长期储存和运输氢的可行性;德国Hydrogenious Technologies 公司利用二苄基甲苯储氢体系,将LOHC 撬装脱氢设备部署到加氢站,实现从绿色制氢到交通运输的示范;2023 年,德国Hydrogenious Technologies 公司实施了200 kW 的LOHC/燃料电池示范项目,推进船上应用零排放技术,中国化学建投公司与武汉氢阳能源控股有限公司进行了全球首套常温常压LOHC 加注一体化装置示范。虽然LOHC 技术已有示范应用,但仍存在很多亟待解决的技术难题。如有机氢载体加(脱)氢反应过程缓慢,环烷烃类等储氢载体脱氢温度高,副反应并发,释放氢气纯度不足,反应过程严重依赖贵金属催化剂,大大增加了LOHC 技术的经济成本。目前低碳能源面临着区域限制、季节限制问题,LOHC载体与现有的油气储运设施适配度高,可大规模、远距离运输,在能量丰富的时间和地点装载LOHC 化合物,然后运送到需要的地方使用,在空间和时间上实现了能源生产和能源使用的脱钩[11-14],因此可以预期随着LOHC关键技术的突破,将引起氢能储运方式的变革。
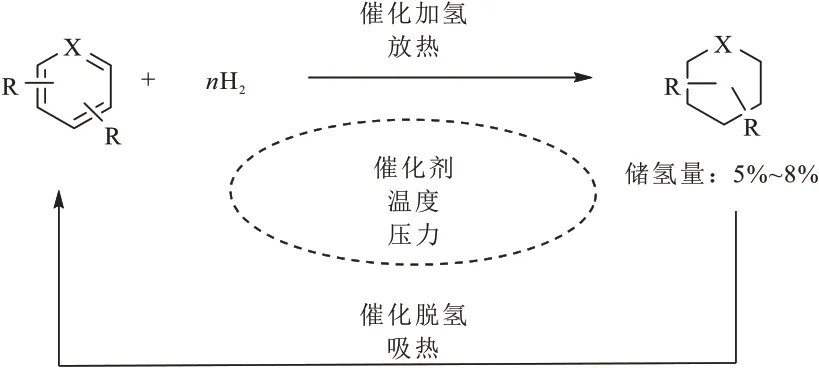
图1 LOHC技术原理示意图Fig.1 Principle schematic diagram of LOHC technology
本文首先对常见的有机氢载体进行分类,并分析各有机氢载体的物理化学性质以及在加(脱)氢反应上的特征差异;然后从加(脱)氢反应机理和催化剂的研究等方面对LOHC体系的现状进行总结;最后对LOHC技术的发展前景进行了展望。
1 有机氢载体加(脱)氢过程研究进展
在过去几十年中,研究最多的有机氢载体包括环烷烃类、杂芳族类以及醇胺类化合物,除此之外,还有对有机化合物进行改性后的金属有机氢载体。本文将LOHC技术以不同储氢载体进行了分类,给出了各储氢载体作为有机氢载体的关键参数和理论储氢量,还探索了低共熔混合储氢体系。从加(脱)氢反应焓、加(脱)氢反应路径和速率限制步骤等方面总结了不同有机氢载体加(脱)氢反应热力学、动力学特性,同时提出了各有机氢载体加(脱)氢反应过程的特征差异,以便更深入地了解不同有机氢载体在有LOHC技术中的应用。
1.1 环烷烃类有机氢载体储/放氢过程
环烷烃类化合物是最早被用作液态有机储氢分子的一类有机化合物[15-18],这与环烷烃类化合物储氢量高、原料易得且熔沸点区间合适等优点相关。在环烷烃类化合物中,针对甲苯/甲基环己烷储氢系体、二苄基甲苯/全氢化二苄基甲苯储氢体系的研究较为成熟,目前已有示范应用的报道。虽然芳烃的催化加(脱)氢化反应在化工生产中很常见,但是运用到储氢体系中还是存在诸多障碍。如脱氢温度高、能耗高,这对某些应用终端(如燃料电池)来说是较大的负担;高温条件下容易发生氢裂解等副反应,从而导致催化剂结焦失活、催化脱氢反应选择性不高和产物氢气不纯等问题;环烷烃类化合物储氢体系在反应温度下容易挥发,尽管整个氢化反应可以在密闭空间里进行,但需要额外增加一个氢气分离步骤,这增加了LOHC技术的推广应用难度[19-23]。因此有学者提出用脱氢焓低的其他有机化合物作为新型储氢载体,其中杂芳族化合物是典型代表。
1.2 杂芳族类有机氢载体储/放氢过程
PEZ等[24]经过理论计算,预测了不同环数的Π-共轭化合物的标准氢化焓,发现在碳环上引入杂原子,特别是氮原子,化合物的标准氢化焓会显著降低。应用于氢气储存的Π-共轭化合物应具有如下结构:(1)含氮稠环化合物:即含一个氮原子的单杂环化合物(五元环、六元环)与至少一个苯环稠和形成的化合物,如咔唑、N-烷基咔唑和N-烷基吲哚等;(2)含氮原子的单环芳香化合物:即含有一个或多个不相邻氮杂原子的单环芳香化合物,如吡啶。吡啶的标准氢化焓为62.76 kJ/mol,比苯加氢低了5.86 kJ/mol。此外,PEZ等还指出,氮原子掺杂在五元环中的含氮稠环化合物的氢化热最低;当环上引入烷基取代基时,含氮稠环化合物的标准氢化焓会轻微降低;而带有烷基取代基的含氮稠环化合物,熔点低、沸点高,更能满足LOHC 要求。总的来说,将含氮杂环化合物作为有机氢载体,储放氢反应条件更温和。许多学者以此为切入点,对这类新型LOHC分子开展了一系列实验研究。
1.2.1 咔唑类有机氢载体储/放氢过程
咔唑由两个苯环并一个吡咯环组成,含氮杂环是吡咯,氢化热低,而且含有两个苯环,大大增加了储氢密度。但是咔唑本身熔点很高,在常温常压下为固态,限制了其作为有机氢载体的使用范围。当在氮原子上引入取代基(如NEC 和N-丙基咔唑(NPCZ))后,新化合物熔点显著降低,同时可以保持较低的氢化热。咔唑类化合物有机氢载体的关键参数见表2。NEC和NPCZ能在200 °C以下进行可逆储/放氢反应,同时还具有良好的循环稳定性。NEC 在10 次循环充放氢实验后,储氢量下降不到2%[25],NPCZ在5次可逆储氢实验后,储氢量下降不到1%[26]。NEC和NPCZ在脱氢反应过程中,没有副产物生成,氢气纯度可达99.99%,被认为是继环烷烃类化合物后较有应用前景的LOHC技术载体。
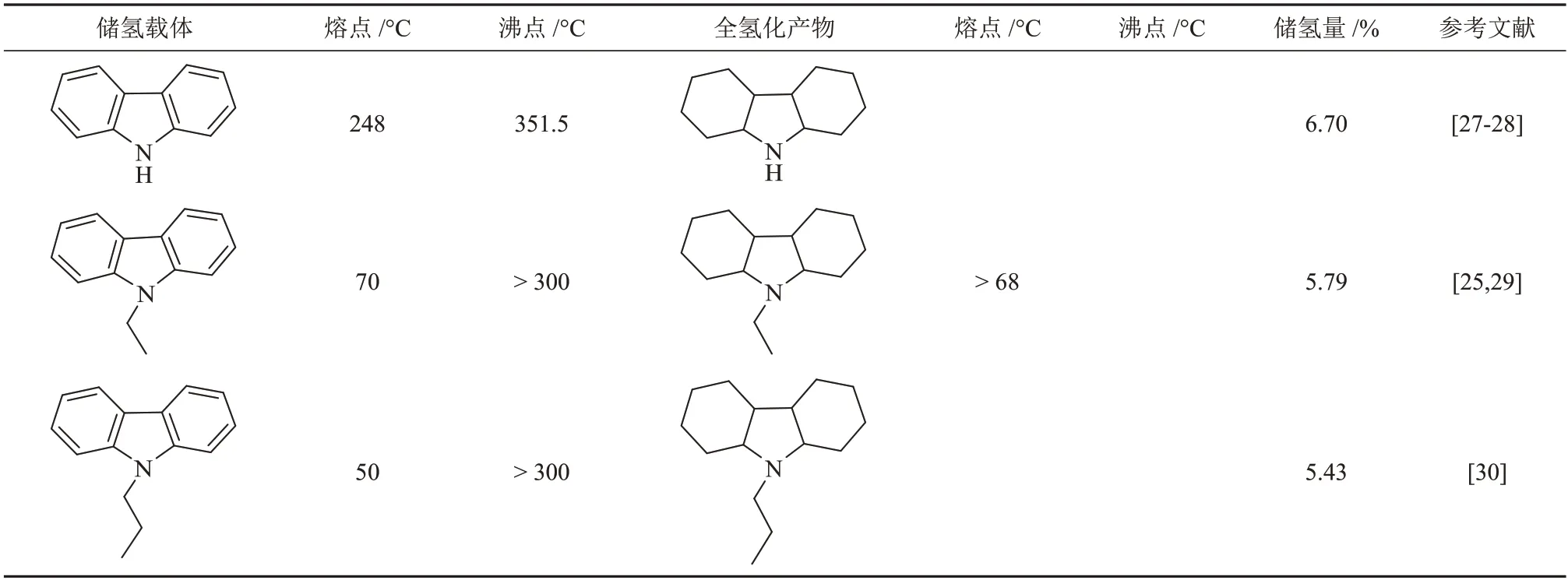
表2 咔唑类化合物有机储氢载体及对应全氢化产物的关键参数Table 2 Key parameters of carbazole compound organic hydrogen storage carriers and corresponding fully hydrogenated products
SOTOODEH 等[31]以NEC 为氢载体进行了储(放)氢研究,结果表明,NEC的加氢反应首先在苯环上发生,然后在含氮杂环上发生,温度对NEC加氢反应的反应速率有较大影响。当温度从130 °C升高到150 °C时,NEC的加氢反应速率常数从0.0121 min-1增加到 0.0492 min-1,1 g催化剂作用下的初始反应速率从3.60 mmol/min增加到14.66 mmol/min,随着反应温度的升高,NEC的加氢反应速率逐渐加快。此外,温度对全氢化NEC脱氢反应的产物分布也有较大影响。YANG等[25]利用Pd负载量为5%(质量分数)的Pd/Al2O3催化了12H-NEC 的脱氢反应过程,发现在128 °C 和145 °C 分别只能得到8H-NEC和4H-NEC中间体,脱氢温度高于178 °C则可以得到NEC。实验结果表明,12H-NEC的脱氢反应经历了12H-NEC →8H-NEC、8H-NEC → 4H-NEC和4H-NEC → NEC 这3步过程,各步的脱氢反应速率常数分别为0.321 min-1、0.187 min-1和0.002 min-1,4H-NEC向NEC的转化是脱氢反应的决速步骤。这也解释了当脱氢反应温度低于178 °C时,氢气释放不彻底的原因。NEC的具体加(脱)氢反应路径见图2。
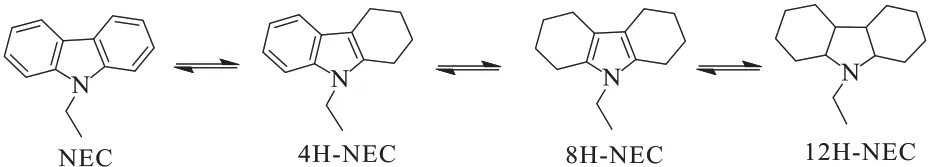
图2 NEC的加(脱)氢反应路径[25,31]Fig.2 Hydrogen addition (dehydrogenation) reaction route of NEC[25,31]
NPCZ与NEC的加(脱)氢反应过程类似,全氢化NPCZ的脱氢反应过程也是含氮杂环先进行脱氢,接着是六元环脱氢,具体反应路径为12H-NPCZ →8H-NPCZ → 4H-NPCZ → NPCZ。DONG 等[26]对这3 个反应阶段的动力学进行了详细研究,确定了NPCZ的脱氢反应为一级反应,每一步的表观活化能分别为90.0 kJ/mol、90.6 kJ/mol和96.4 kJ/mol。此外,脱氢反应温度越高,全氢化NPCZ的脱氢反应速率越快,在5%Pd/Al2O3催化作用下,于180 °C反应360 min的转化率为90%,190 ℃反应300 min 的转化率为100%,200 °C反应240 min能获得100%的转化率。
1.2.2 吲哚类有机氢载体储/放氢过程
吲哚是一类具有苯并五元含氮吡咯环结构的富电子化合物,具有大的Π-共轭体系和刚性稠环结构。吲哚类化合物的标准氢化焓低于62.76 kJ/mol,其储氢性能也引起了诸多关注。表3列出了部分吲哚类储氢分子作为有机氢载体的关键参数。特别的是,7-乙基吲哚(7-EⅠD)、8H-7-乙基吲哚(8H-EⅠD)、N-甲基吲哚和8H-N-甲基吲哚的熔点较低,在常温常压下呈液态,相对于卡唑类化合物具有一定的储运优势。
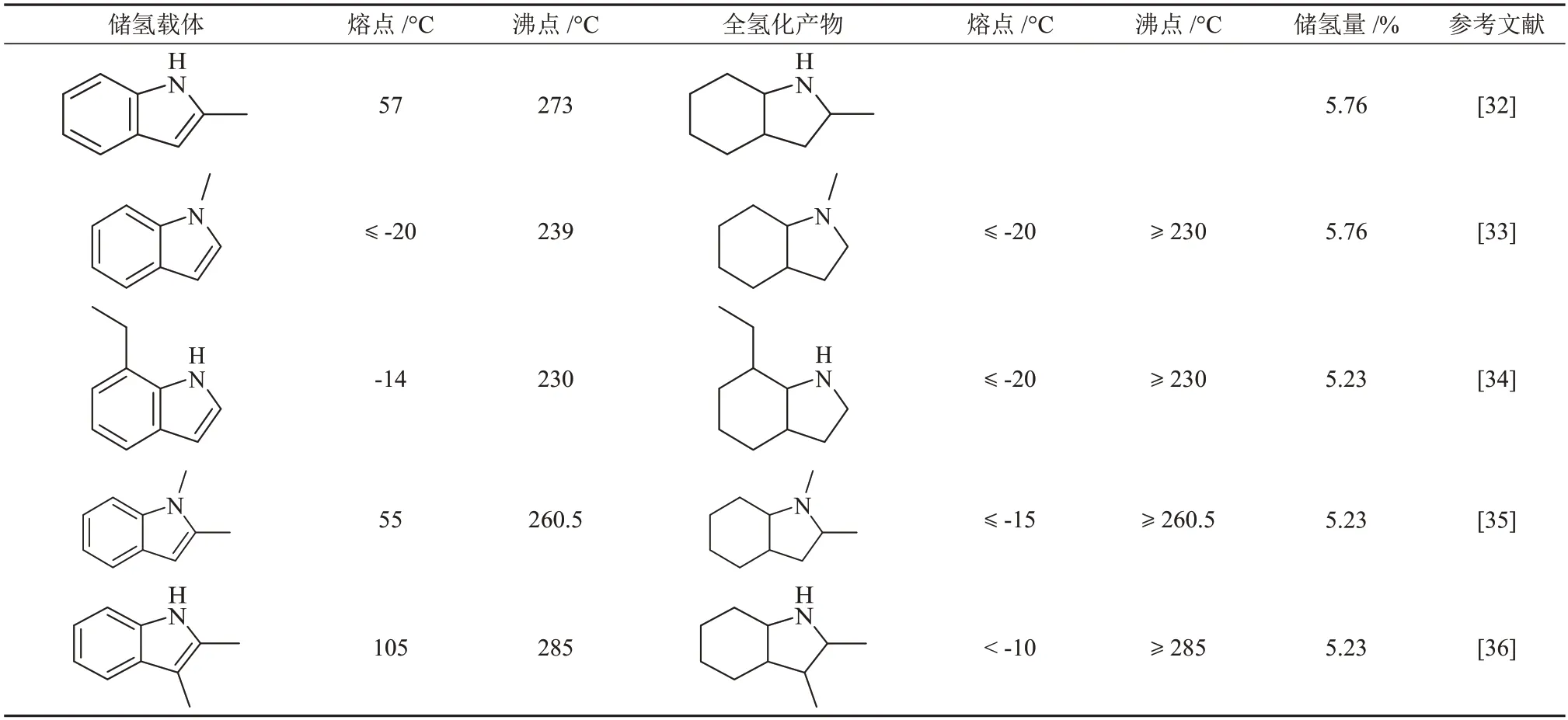
表3 吲哚类化合物储氢载体及对应全氢化产物的关键参数Table 3 Key parameters of indoles compound organic hydrogen storage carriers and corresponding fully hydrogenated products
程寒松课题组[32,35-38]系统地研究了取代基位置、数量和烷基链长度对吲哚类化合物加氢和脱氢反应动力学的影响,结果见表4。不同烷基取代的吲哚衍生物加(脱)氢反应动力学差异显著。与2,3-二甲基吲哚相比,2-甲基吲哚脱氢温度更低,反应速度更快。在相同催化剂的催化作用下,2-甲基吲哚在160 °C 下反应40 min 的转化率为100%,8H-2-甲基吲哚在190 °C下反应240 min的转化率为100%,加氢和脱氢反应的表观活化能分别是21.0 kJ/mol和27.1 kJ/mol。2,3-二甲基吲哚在190 °C下反应240 min的转化率为100%,8H-2,3-二甲基吲哚在210 °C 下反应300 min的转化率为93.6%,加氢和脱氢反应的表观活化能分别为85.1 kJ/mol 和111.9 kJ/mol。研究人员认为这种动力学差异可能与取代基的空间位阻效应有关,烷基取代基越多,空间位阻越大,对应的加氢和脱氢反应的动力学性质越差。对N 上的取代基而言,烷基1-位取代化合物比其他位取代化合物的脱氢反应的表观活化能高,如N-甲基吲哚和1,2-二甲基吲哚的脱氢反应表观活化能分别为122.99 kJ/mol 和111.90 kJ/mol,远高于2-甲基吲哚、7-乙基吲哚和2,3-二甲基吲哚的脱氢反应表观活化能(依次为27.1 kJ/mol、101.9 kJ/mol和39.6 kJ/mol)。
吲哚类化合物的加(脱)氢反应路径与咔唑类化合物类似。加氢反应时,通常是苯环先部分加氢,然后含氮杂环加氢。脱氢反应时,则先从含氮杂环开始脱氢。7-乙基吲哚的加(脱)氢反应路径见图3。
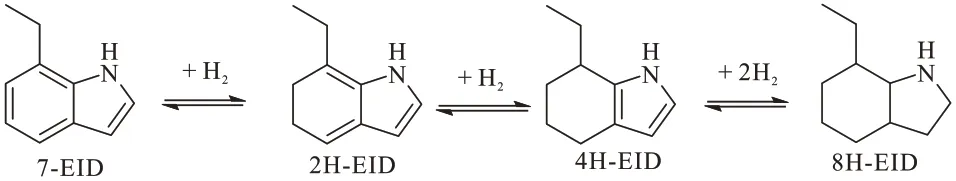
图3 7-乙基吲哚的加(脱)氢反应路径[34]Fig.3 Hydrogen addition (dehydrogenation) reaction route of 7-ethyl indole[34]
目前,大多数表观活化能较小的氮杂环化合物,如2-甲基吲哚、NEC和NPCZ等在常温常压下为固态,然而理想的有机储氢体系应该是由全液态有机化合物组成。CHEN 等[39]将2-甲基吲哚、NEC 和NPCZ以质量分数40%、24%和36%混合(质量储氢密度为5.64%),组成了熔点为25 °C 的低共熔储氢体系,并利用该课题组开发的PdO/Zr-C3N4催化剂,在140 °C下实现了混合储氢载体完全脱氢,完全脱氢产物的收率达到94.4%。这种将不同类型LOHC载体按适当比例混合组成低共熔储氢体系的策略为LOHC技术中的氢载体研究提供了新思路。
1.2.3 其他杂环类有机氢载体储放氢过程
除上述研究较多的咔唑、吲哚类含氮杂环储氢载体,近年来科研人员为了提高储氢量,还拓展了理论储氢量高的其他杂环类储氢载体。如含氮杂环化合物吖啶(ACD)、喹啉和吩嗪,以及含硫杂环化合物噻吩和苯并噻吩等。这些杂环类化合物有机氢载体的关键参数见表5。杂环类化合物因不同苯并杂环而呈现不同的储氢性能,在此分析了苯并五元杂环、苯并六元杂环和含硫杂环化合物作为有机氢载体的储氢性能,同时分析了氮原子掺杂数量、苯环数等因素对有机氢载体储氢性能的影响。
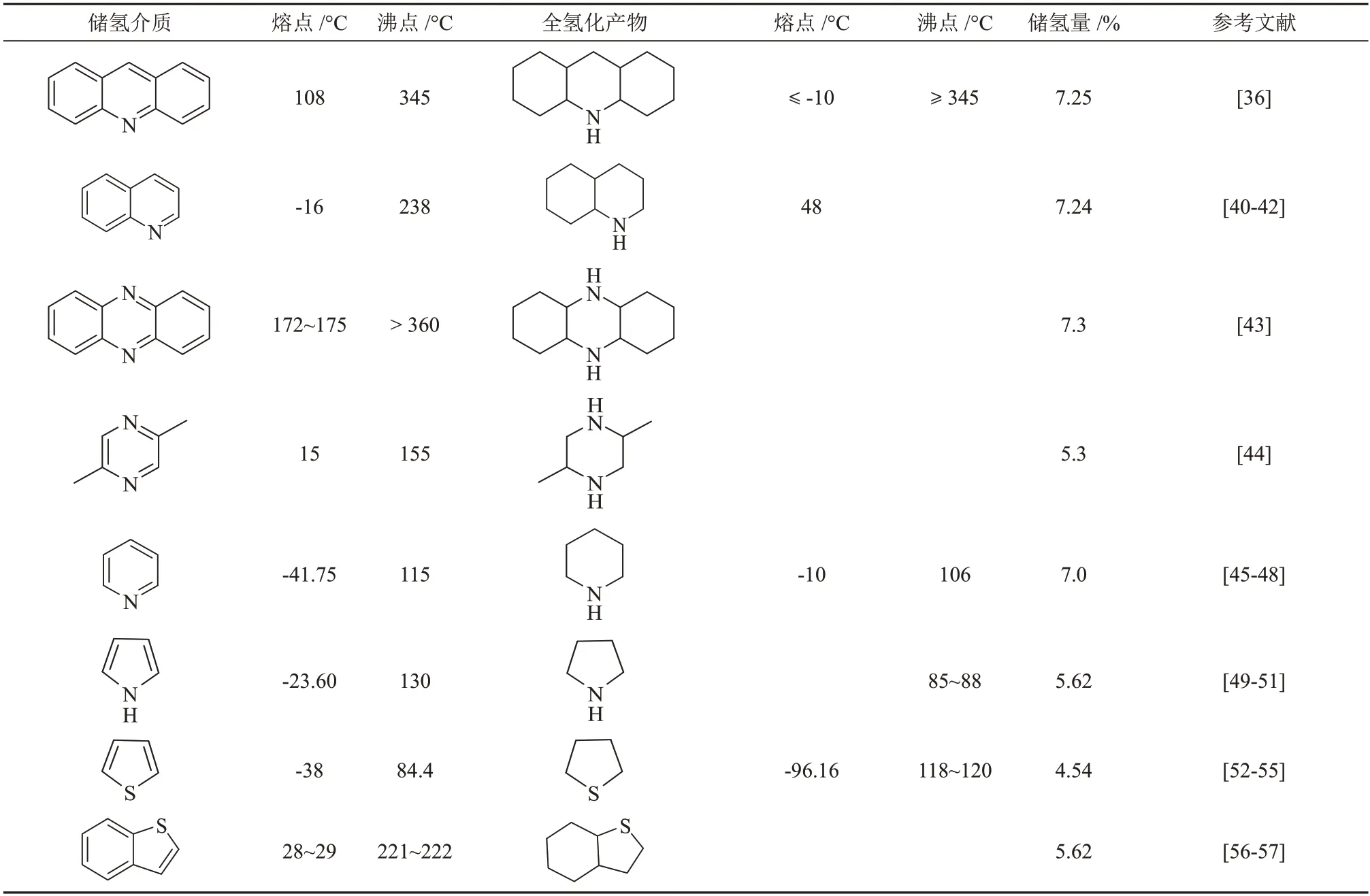
表5 其他杂环类化合物储氢载体及对应全氢化产物的关键参数Table 5 Key parameters of other heterocyclic compound hydrogen storage carriers and corresponding fully hydrogenated products
苯并六元含氮稠环化合物如吖啶、喹啉与苯并五元含氮稠环化合物N-烷基咔唑、N-烷基吲哚作为储氢载体时的加(脱)反应热力学、动力学存在明显的区别,但同时也有相似的反应路径和速率限制步骤。YANG 等[58]利用5%Ru/Al2O3作为催化剂,在140 °C、8 MPa氢气条件下实现了ACD的完全氢化。在反应初期,ACD同时转化为中间体4H-ACD、8HACD和14H-ACD,其中中间体8H-ACD的浓度高于其他中间体,随着反应时间的增加,中间体4HACD、8H-ACD 进一步转化为14H-ACD,其中8HACD向14H-ACD的转化是氢化反应的速率限制步骤。喹啉类化合物氢化反应条件苛刻,在目前已报道的文献[59-60]中,氢化产物以1,2,3,4-四氢喹啉为主,很难进一步生成十氢喹啉,这可能与苯并六元含氮杂环的电子效应更强、空间位阻更大有关,因此导致很难氢化碳环上的氢。总的来说,吖啶、喹啉的氢化反应温度和压力均比N-烷基咔唑高,但加(脱)氢反应路径相似且都是多步反应过程中最后一步为速率限制步骤。
含多个氮原子的含氮杂环化合物比含单个氮原子的含氮杂环化合物的加(脱)氢反应更容易进行。目前已有无溶剂条件下含多个氮原子的含氮杂环化合物加(脱)反应的研究报道。FUJⅠTA 等[44]以2,5-二甲基吡嗪为储氢载体,以铱络合物为催化剂,在相对较低的氢气压力(0.15 MPa)和反应温度(138 °C)下实现了无溶剂条件下的2,5-二甲基吡嗪氢化反应和有微量溶剂(H/S值大于30 mmol/mL,H指释放或储存氢气的物质的量(mmol),S指溶剂体积(mL))存在的2,5-二甲基哌嗪的脱氢反应,极大地提高了整个体系的储氢量,具体反应如图4所示。综合以上研究结果可知,溶剂对可逆储氢反应的影响很大,而无溶剂反应条件最为苛刻。H/S值可用于评估每个系统中使用的溶剂量,H/S值越大,意味着反应过程需要的溶剂量越少,反应难度也会增加。
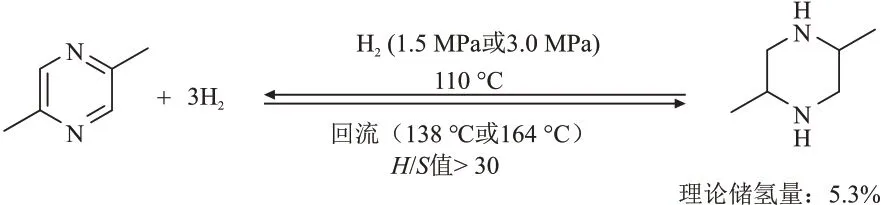
图4 2,5-二甲基吡嗪的加(脱)氢反应[44]Fig.4 Reaction of hydrogen addition (dehydrogenation) of 2,5-dimethylpyrazine[44]
稠环含氮化合物理论储氢量高,但是在其加(脱)氢反应过程中会生成多个反应中间体,加氢或氢气释放不彻底,反应收率较低,整体的加(脱)氢反应循环率比单杂环含氮化合物低。FORBERG等[61]设计合成了具有多个活性位点的Pd2Ru@SiCN双金属催化剂,并成功实现吩嗪(Phen)加氢和14H-吩嗪(14H-Phen)脱氢的多次循环反应。这也是在LOHC储氢技术中,迄今为止储氢量最高的有机氢载体可逆加脱氢循环次数,具体反应如图5所示。吩嗪的储放氢反应过程相对复杂,7次循环放氢的结果显示,氢化产物主要以14H-Phen 为主,8H-Phen 占比低于17%(质量分数),4H-Phen 占比低于4%,脱氢产物主要是Phen,4H-Phen占比低于6%,8H-Phen占比低于23%。总的来说,在这7次循环中,氢化产物和脱氢产物分布都出现了一些微小的变化,储氢量保持在5.81%~7.08%,存在多次循环储放氢反应后储氢量降低的问题。
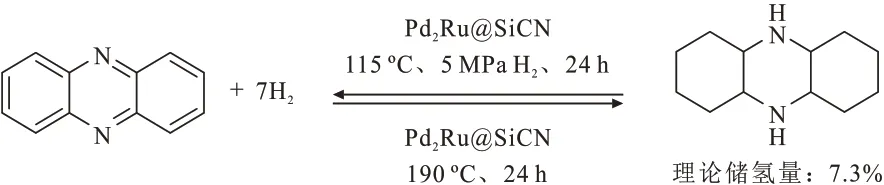
图5 吩嗪的加(脱)氢反应[61]Fig.5 Reaction of hydrogen addition (dehydrogenation) of phenazine[61]
FANG 等[62]利用氧化镁负载的Ru 纳米颗粒(5%Ru/MgO)对多种芳香族化合物的催化加氢反应进行了深入研究,认为吡啶类化合物较吡咯类化合物更容易氢化。在150 °C、5 MPa 的条件下,吡咯的和吡啶的反应速率分别为为1.60 mol/(L·h)和1.96 mol/(L·h)。与单杂环相比,稠环杂环更难氢化,吡咯和吡啶可以被完全还原为吡咯烷和哌啶,而稠环杂环则生成不完全氢化的混合产物。喹啉主要在氮杂环上氢化,只有一小部分产物来自碳环的氢化,吖啶的氢化在碳环和N-杂环产物都有生成。含硫杂环化合物在进行催化氢化反应时,脱硫产物容易使催化剂中毒。
总的来说,苯并五元环的含氮化合物比苯并六元环的含氮化合物更容易加(脱)氢。氮原子掺杂数量对储氢载体标准氢化焓有影响,含多个氮原子的化合物氢化焓更低,储氢性能更优。含氮稠环化合物虽然比单杂环含氮化合物理论储氢量高,但是存在反应过程复杂、生成中间体较多、加氢或释氢不彻底和循环储氢率低等问题。
1.3 金属有机氢化物有机氢载体
中国科学院大连化学物理研究所何腾、陈萍团队提出利用具有不同电负性的金属来调变有机储氢材料的电子性质,从而调变材料的脱氢焓的策略,首次开发出了一种全新的改性有机无机杂化储氢材料——金属有机氢化物。金属有机氢化物由金属阳离子和有机阴离子组成,种类丰富,性质各有不同。该团队共设计预测了近100种金属有机氢化物的热力学性质,包括苯酚钠[63]、咔唑锂[64]和吲哚锂[65]等。根据储氢量大于5.0%且反应脱氢焓变介于25~35 kJ/mol 之间的要求,研究人员从中筛选出了20 余种具有应用前景的储氢材料。理论计算结果表明,引入金属的供电子能力越强,材料的脱氢焓值越低。
以金属(Li、Na、K、Ga 和Mg)修饰的苯酚/环己醇为例[63],理论计算结果表明,脱氢焓值由高到低依次为:C6H11OH、(C6H11O)2Mg、C6H11O)2Ga、C6H11OLi、C6H11ONa和C6H11OK,随着金属给电子能力增强,材料的脱氢焓变逐渐降低。苯酚钠-环己醇钠的脱氢焓变可以从64.5 kJ/mol降低到50.4 kJ/mol。进一步的实验研究证明,在商业催化剂5%Ru/Al2O3作用下,苯酚钠-环己醇钠可以在低至30 °C 的温度下实现加氢反应。对于催化脱氢反应,苯酚钠-环己醇钠固态体系可以在5%Pd/Al2O3催化作用下,在140 °C条件下脱氢,而将材料溶于水后,脱氢温度可以降低到100 °C。
在金属修饰的苯酚/环己醇研究成果上,TAN等[64]进一步提出了利用碱金属/碱土金属改性氮杂环储氢材料,开创了一类全新的金属氮杂环化合物。通过理论计算,得出了金属咔唑盐的脱氢焓值为26.4~40.0 kJ/mol,以脱氢焓值(33.7 kJ/mol)与理想脱氢焓值(30.0 kJ/mol)十分接近且储氢量最优的咔唑锂(6.48%)为例进行了储氢性能测试。咔唑锂(Li-CZ)可以在Ru基催化剂作用下,在100 °C和7 MPa氢压下实现完全氢化。而在全氢化咔唑锂(Li-12H-CZ)脱氢过程中,Ru基催化剂的催化作用不明显,而Pd/C和Rh/Al2O3可以在200 °C下将Li-12H-CZ部分转化为Li-CZ(转化率分别为72%和44%)。在相同的实验条件下,十二氢咔唑完全没有反应。吲哚锂(Li-ⅠD)[65]的可逆储氢反应与咔唑锂相类似,可以实现Li-ⅠD 到全氢化吲哚锂(8H-Li-ⅠD)的完全转化,但是脱氢反应只得到54.1%的转化率。作者认为Li-12H-CZ 和8H-Li-ⅠD 在脱氢反应中呈浆状,非均相催化剂在此条件下的传质阻力被放大,导致催化脱氢反应转化率不高。苯酚钠、Li-CZ 和Li-ⅠD 的加(脱)氢反应见图6。
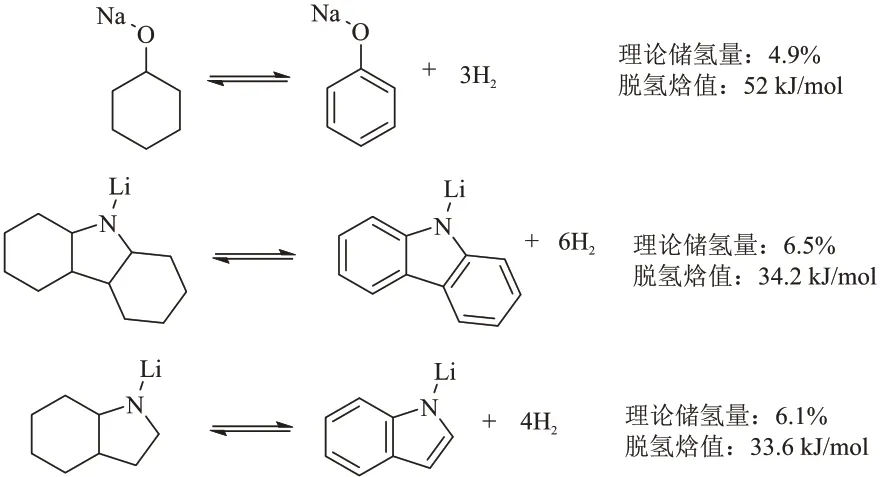
图6 苯酚钠、咔唑锂和吲哚锂的加(脱)氢反应[63-65]Fig.6 Reaction of sodium phenol,lithium carbazole and lithium indoline in hydrogen addition (dehydrogenation)[63-65]
金属有机氢化物储氢材料克服了金属氢化物可逆储氢容量衰减严重和一般的有机液体化合物脱氢温度高的问题,兼有储氢容量高和适宜热力学的优点。此外,有机物种类多样,与无机金属杂化后可以衍生出更多种类的储氢材料,该研究为未来低温可逆储氢材料的开发开辟了崭新的思路,是一类具有应用前景的储氢体系。
1.4 其他LOHC载体
HU等[66]、SHAO等[67]和KUMAR等[68]通过醇-胺脱氢偶联酰胺化反应和酰胺的氢化反应实现氢气的释放和存储,该类体系使用的储氢载体包括2-氨基乙醇、乙二胺和1,4-丁二醇等,具有原料易得,价格低廉等优点,整个可逆储氢过程可以在温和的氢气压力和低于200 °C 的温度条件下完成。但是目前这类储氢载体的加(脱)氢反应仅有络合物催化剂(如钌络合物)作用下的研究报道。胺类和醇类储氢载体的反应见图7。
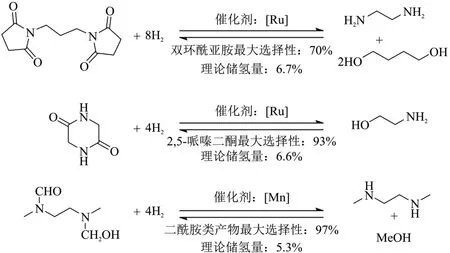
图7 胺类和醇类储氢载体的反应[66-68]Fig.7 Reaction of amines and alcohols hydrogen storage carriers[66-68]
当储氢体系为乙二胺/1,4-二丁醇混合物时[68],在135 °C、0.4 MPa 氢气压力下反应40 h,双环酰亚胺转化率为99%,并且1,4-丁二醇和乙二胺的收率均超过90%。对于脱氢反应,乙二胺和1,4-二丁醇经过脱氢偶联形成双环酰亚胺,在120 °C 下,乙二胺和1,4-二丁醇完全转化但双环酰亚胺收率仅有70%,其余为副产物内酯(10%)和低聚酰胺。此外,作者还研究了乙二胺/1,4-二丁醇与双环酰亚胺之间的相互转化循环。首次循环反应以双环酰亚胺为底物,将氢化反应得到的乙二胺/1,4-丁二醇混合物在120 °C 下脱氢,经第一次循环反应后,双环酰亚胺的收率为68%,收集到氢气78 mL。将双环酰亚胺和副产物的混合物进行第二次加氢/脱氢循环反应,双环酰亚胺的收率为64%,收集到氢气71 mL。从两次相互转化循环实验来看,乙二胺和1,4-二丁醇脱氢反应过程生成的副产物内酯和低聚酰胺是储氢量降低的主要原因。当以2-氨基乙醇作为储氢材料时[66],该体系最大储氢量为6.6%,与乙二胺/1,4-丁二醇脱氢反应有副产物生成的情况类似,2-氨基乙醇在经过分子间环状脱氢反应生成终产物哌嗪-2,5-二酮(甘氨酸酐,GA)的同时也会有少量的线性肽副产物生成。
近期,LⅠU 等[67]开发了一种廉价金属锰的新型催化剂,成功催化了以N,N-二甲基乙二胺和甲醇混合物作为储氢载体的可逆储氢反应。对比来看,虽然锰络合物催化剂进行加(脱)氢反应时的反应条件比贵金属催化剂更加苛刻,但是在此储氢体系中,N,N-二甲基乙二胺和甲醇的脱氢反应在165 °C下的转化率为100%,其中目标产物二酰胺类产物的收率达到94%,选择性高达97%。同时在110 °C、0.6 MPa氢气条件下二酰胺类产物也能以95%的高转化率还原成N,N-二甲基乙二胺和甲醇。基于锰催化的N,N-二甲基乙二胺和甲醇储氢体系理论储氢量为5.3%,虽然储氢量略低于2020 年美国能源部发布的储氢量指标(5.5%),但是该催化体系转化率、选择性高,几乎无副产物生成,氢气纯度得到了保证。
综上所述,在这些有机氢载体中,环烷烃类化合物储氢量高,在常温常压下呈液体状态,但是这类化合物存在脱氢反应温度高的问题。含氮杂环咔唑、吲哚类储氢载体可以在较低温度(150~200 °C)下进行可逆储氢反应,显示出优异的热力学、动力学性质。特别的是,N-甲基吲哚储氢体系在常温常压下的熔点低于-20 °C,理论储氢量为5.76%,且可在较为温和的实验条件下实现可逆储放氢循环,在一定程度上促进了N-杂环类氢载体在LOHC 技术中的应用[69-72]。吖啶、吩嗪等储氢载体的理论储氢量高达7%,但是存在加(脱)氢反应过程复杂,实际循环储氢率低等问题。氨、醇和酰胺作为一类比较新颖的储氢体系,原料廉价易得,脱氢反应温度低于200 °C,但是反应选择性高度依赖催化剂的使用。以苯酚钠-环己醇钠为代表的金属有机储氢体系展示了更低的脱氢焓,为未来开发低温可逆储氢材料开辟了新思路。
2 LOHC加(脱)氢催化剂研究进展
有机氢载体的催化加(脱)氢反应是一个逐步而缓慢的复杂反应过程,会生成较多反应中间体以及加氢终产物包含多个立体异构体。此外有机氢载体的脱氢反应是体积增大的吸热反应,在一定压力条件下,反应温度越高,反应越容易进行,而加氢反应则是在高压低温条件下更容易进行。加快反应速率主要靠加大氢气压力或一定程度上的升温。因此如何从催化剂角度提高有机氢载体加(脱)氢反应的深度以及反应速率是目前催化剂设计面临的一大挑战。有机氢载体催化剂的催化性能受很多因素的影响,如催化剂载体、金属活性组分的种类和催化剂的结构敏感性等。
2.1 LOHC加氢催化剂
目前已经开发的非均相有机氢载体储氢催化剂有很多种,如Pt、Pd、Rh和Ru等贵金属催化剂[79]。在实际实验研究过程中,新型含氮杂环如NEC等有机氢载体的催化加氢反应完全可以在贵金属的催化作用下完成,比如常用的Ru基催化剂。
WU 等[73]将一种稀土氢化物负载的钌催化剂(Ru/YH3)用于N-杂环储氢载体的氢化反应。Ru/YH3采用改性化学气相沉积法合成,虽然YH3的比表面积小,在化学气相沉积过程中Ru损失较多,比采用相同方法合成的Ru/Al2O3Ru负载量低3%,但是Ru/YH3的转化数却是Ru/Al2O3的3 倍,这也说明金属负载量对催化剂催化活性的影响有限,载体上的化学环境也会影响金属活性组分的催化性能。Ru/YH3对NEC的全氢化产物具有高立体选择性,在150 °C、3 MPa氢气条件下,产物中全顺式结构12H-NEC 占比91%,而这是其他催化剂载体负载的Ru基催化剂不具备的。同时也有研究表明,与其他全氢化产物相比,全顺式结构的12H-NEC能更快地进行催化脱氢反应[30]。经实验证明,Ru/YH3的特殊催化活性源于一种新型的氢转移方式,即通过YH3可逆的吸氢和解吸实现从H2到NEC 的氢转移,从而使Ru/YH3在低金属负载量的情况下也具有高催化性能。
GE 等[74]研究报道了一种Ru 单原子和分子筛(Beta)协同催化促进NEC 催化低温加氢反应的Ru(Na)/Beta 高效催化剂。与传统的Ru/Al2O3催化剂相比,单原子Ru(Na)/Beta 催化剂具有适宜的酸性,分散的Ruδ+位点和相邻的酸位点可以协同活化H2分子,在较低温度下快速促进NEC 加氢。在100 °C 下,1.5 h 时的吸氢率为5.69%(质量分数),NEC 转化率为99.19%,达到NEC 理论储氢量的98.28%。借助各种表征技术,GE 等发现单原子分散的Ru(Na)/Beta 具有较高的异裂活化H2分子的能力和氢溢流能力。此外,作者还发现使用氢氧化钠为沉淀剂的Ru(Na)/Beta 催化剂比未添加氢氧化钠沉淀剂的Ru(Na)/Beta催化剂活性高,猜测这可能是碱金属离子(Na+)具有抑制Ru(Na)上的Ruδ+还原及团聚的作用。
2.2 LOHC脱氢催化剂
目前LOHC 的技术难点在于催化脱氢反应,以NEC为例,在160~190 °C、6~9 MPa反应条件下,NEC的氢化反应在较短的时间内能完全生成12H-NEC,相反,12H-NEC在温和的条件下很难完全脱氢。因此开发高效的脱氢催化剂是LOHC 研究领域的热点[75]。
YANG 等[76]以三氧化二铝为载体,研究了Pd、Pt、Ru 和Rh 等贵金属活性组分对12H-NEC 催化效率的影响。反应1 h 内,金属催化剂的催化活性由高到低依次为:Pd、Pt、Ru 和Rh。其中Pd 和Pt 催化剂分别在240 min 和300 min 后可以实现12H-NEC的完全脱氢。Ru催化体系的最终产物由71.46%的NEC和28.54%的4H-NEC组成。而在基于Rh催化剂的反应中,全脱氢产物的NEC 只有10.64%,不完全脱氢产物则高达89.36%。WANG 等[77]以提高脱氢终产物NEC的选择性为出发点,采用功能性载体还原氧化石墨烯(rGO),设计制备了一系列单金属M/rGO 催化剂(M=Pd、Pt、Ru、Rh 或Au)。结果表明,催化性能由高到低依次为:Pd/rGO、Pt/rGO、Rh/rGO、Ru/rGO和Au/rGO。其中Pd/rGO催化剂实现了12H-NEC在180 °C下7 h内的完全脱氢,NEC的选择性高达97.65%,而市售催化剂5%Pd/Al2O3在相同条件下对NEC 的选择性仅有44.77%。从上述实验研究中可以得出,在众多贵金属催化剂中,Pd 催化剂在12H-NEC 脱氢反应中表现出最优的催化活性。
接着,WANG等[78]通过引入廉价金属Cu,构筑了rGO负载的双金属PdCu合金型催化剂。Pd1.2Cu/rGO催化剂在贵金属用量降低了20%的前提下保持了Pd/rGO 催化剂的脱氢反应效果,在180 °C 下,反应1 h 后,12H-NEC 的转化率达到100%,7 h 后实现了100%NEC的生成。作者分析了双金属PdCu催化剂中微观电子结构随PdCu 纳米粒子不同配比的变化规律,进而建立了双金属PdCu 催化剂催化活性与PdCu纳米粒子微观电子结构间的关联,最终在降低贵金属用量的情况下,充分利用双金属催化剂的协同催化效应使PdCu/rGO达到最优催化性能。
Pd 基多相催化剂在催化12H-NEC 脱氢反应中具有较高的催化活性。然而,Pd基多相催化剂催化12H-NEC定向多步脱氢反应的催化机理未能被定性定量分析。DONG等[79]利用6组不同Pd负载量的纳米金刚石(Pd/ND)催化剂催化12H-NEC和4H-NEC的脱氢反应,分别探究了Pd纳米颗粒、Pd纳米团簇和Pd单原子对催化活化12H-NEC中特定化学键的贡献。实验结果验证了12H-NEC 的多步脱氢反应需要在具有金属原子系综的多个催化活性位点存在条件下才能完成。其中全暴露Pd 纳米团簇表现出最优的催化效果,既能同时活化多个C—H键和H2分子而且对产物的吸附力不强。该研究为设计经济高效的贵金属催化剂以及催化机理研究提供了思路。
研究人员对碳基无金属催化剂在有机液体氢载体的催化加(脱)氢反应中的应用也有研究。HU等[80]合成了一系列石墨氮组合碳材料(NCs)。在150 °C和常压N2条件下,实现了无金属催化1,2,3,4-四氢喹啉脱氢转化为喹啉的反应。机理研究表明,NCs 的催化活性位源自前驱体热解自组装形成的石墨氮组合结构(CGNs 位点)。CGNs 位点可活化含氮杂环,并在催化剂表面形成不稳定的C—H 键,随后,这种表面活泼氢经重组、脱附形成H2,使NCs 在无氢受体条件下发挥催化作用。
3 结语与展望
LOHC 技术在大规模、远距离储运氢气方面表现出的经济性远优于其他储氢技术,且运输过程不涉及高温高压,安全性较好。虽然LOHC 技术已有示范应用,但还存在脱氢温度高、释氢速率低、储氢载体循环使用率低及加(脱)氢催化依赖贵金属等缺点。
为发展具有产业化和推广价值的LOHC 系统,应具有但不限于以下要求:(1)所选储氢载体在常温常压下呈液态,在运输方面可以适配现有油气储运基础设施;(2)质量储氢密度大于5%;(3)储氢载体在长时间可逆储氢反应中化学结构稳定,不易分解;(4)储氢载体的沸点高,不易挥发;(5)脱氢产生的氢气纯度高,不需纯化即可满足终端用氢要求;(6)储氢载体无毒(低毒)且成本可控。针对以上要求,未来LOHC体系的研究方向为:(1)继续探索可大规模推广的储氢体系;(2)通过调整金属活性组分的化学环境来激活特定低能途径,进一步降低加(脱)氢反应的温度、压力及综合能耗;(3)针对不同应用场景,开展不同工艺设计。
