RNA扭转角预测的深度学习方法*
2024-01-06欧秀娟肖奕
欧秀娟 肖奕
(华中科技大学物理学院,武汉 430074)
1 引言
RNA分子三级结构模建是分子生物物理学研究的基本问题之一,对理解RNA的功能和设计新的结构有重要意义[1-3].RNA分子三级结构模建是给出RNA分子的核苷酸序列构建其三级结构[4-10].RNA三级结构可以分为主链结构和侧链结构,主链结构由螺旋区和环区构成,由6个扭转角(α,β,γ,δ,ε,ζ)确定,侧链方向由扭转角χ确定(图1).RNA分子主链和侧链结构还涉及共价键键长和键角,但这些键长和键角会相对平衡位置进行微振动,在生理温度这些参数的变化关于平衡位置对称,影响将相互抵消[11].因此,扭转角被认为是RNA分子三级结构的决定因素,预测这些扭转角可以帮助模建RNA分子的三级结构.
扭转角预测在蛋白质分子三级结构模建中已经有深入的研究.与RNA分子不同,蛋白质分子三级结构主要由主链上的2个扭转角ψ和φ确定.从2007年以来,人们提出了不同的神经网络模型预测扭转角ψ和φ.2007年,Real-SPINE1.0使用一层全连接神经网络预测蛋白质主链ψ角,角度的平均绝对误差(mean absolute error,MAE)为54°[12];2008年,Real-SPINE2.0使用同样神经网络和输入特征,角度标签[0°,180°]不变,[-180°,0°]加上360°做一个平移,同时预测蛋白质主链ψ和φ角,角度的MAE分别为38°和25°[13];2009年,Real-SPINE2.0使用两层全连接网络,ψ和φ角预测精度进一步改进,MAE分别为36°和22°[14];2009年和2012年,SPINE XI和SPINE X使用多步神经网络,ψ角预测的MAE分别为33°[15]和35°[16];2015年SPIDER2使用深度学习3层全连接神经网络预测角度的正弦和余弦值,ψ角预测的MAE降低到30°[17];2017年,SPIDER3使用4层双向LSTM模型使ψ角预测的MAE进一步下降为27°[18];2019年,SPOT-1D使用10层以上的LSTM(long short-term memory)残差网络预测角度的正弦和余弦值,ψ角预测的MAE为23°[19];2020年,使用3层全连接网络,滑动窗口特征,ψ角预测的MAE仅为18°[20].对于RNA分子,2021年,SPOTRNA-1D首次使用1层普通卷积和2层膨胀卷积预测RNA的7个扭转角和2个自定义伪角(η,θ)(图1)的正弦和余弦值,α,β,γ,δ,ε,ζ,χ,η,θ的平均绝对误差分别为43.94°,21.94°,32.98°,14.61°,20.69°,33.27°,19.59°,30.25°和32.91°[21].可以看到,相对于蛋白质分子,RNA分子扭转角预测的精度还有待提高.
本文提出了一种基于时序网络深度学习模型预测RNA分子扭转角的方法1dRNA,分别使用深度残差卷积模型(deep residual CNN,DRCNN)和深度超长短期记忆模型(deep HyperLSTM,DHLSTM)预测RNA分子的7个扭转角和2个伪角,以此分析抓取相邻核苷酸特征的卷积网络和抓取全局核苷酸特征的循环网络,哪种网络更合适扭转角预测问题,并将两个模型的结果和抓取间隔核苷酸特征的SPOT-RNA-1D比较.DRCNN模型基于只能看到相邻核苷酸特征的一维卷积,卷积过程不改变序列长;DHLSTM模型基于能看到全部核苷酸的特征、并能改变常规长短期记忆(LSTM)网络权重共享范式的超LSTM网络.结果表明,本文采用的两个深度学习模型都可以进一步提高RNA分子扭转角的预测精度,不同模型在不同角度上各有优势,δ,ζ,χ,η和θ角的预测更适合卷积网络,β和ε角的预测更适合循环网络,而在α和γ角中,抓取间隔核苷酸的膨胀网络更好.
2 深度学习模型
2.1 深度学习模型
DRCNN模型架构如图2所示,由一个一维卷积层[22]开始,输入通道为4,输出通道为512 (卷积输出通道超参数512比256效果好和1024效果类似),训练批次为8 (本文模型在一张11G显存GTX 1080 Ti显卡上能容下的最大样本数),卷积核为15 (卷积核超参数15比7和30效果好),填充方式为“same”,其他为默认值.初始卷积层之后,是4个残差块的依次叠加(残差块的数目1到6测试显示4个残差块效果最好),每个残差块[23]依次包含: 一维批归一化层BatchNorm1d[24](特征维度为512,添加在卷积网络中,有助于模型训练的稳定,效果比LayerNorm样本归一化要好),ReLU激活函数[25](对本文模型激活函数ReLU比tanh和Leaky ReLu效果好),一维卷积层(输入通道维度为512,输出通道维度为512,卷积窗口一次能看到的核苷酸数目为15,填充方式为“same”,其他为默认值),再一维批归一化层,ReLU激活函数和一维卷积层,最后将此层卷积的输出和残差块的输入相加,相加的结果再输入下一个残差块中,重复4次.数据流出残差块后,经过一个ReLU激活函数(激活函数放在残差块外训练效果更好),一维批归一化层(特征维度为512),dropout层(和全连接层连用,减少网络的过拟合,采样概率0.4,比0.2和0.5效果好),全连接层(输入维度512,输出维度18),tanh激活函数(输出区间在[-1,1],和预测角度的正弦和余弦值区间一致)得到输出.
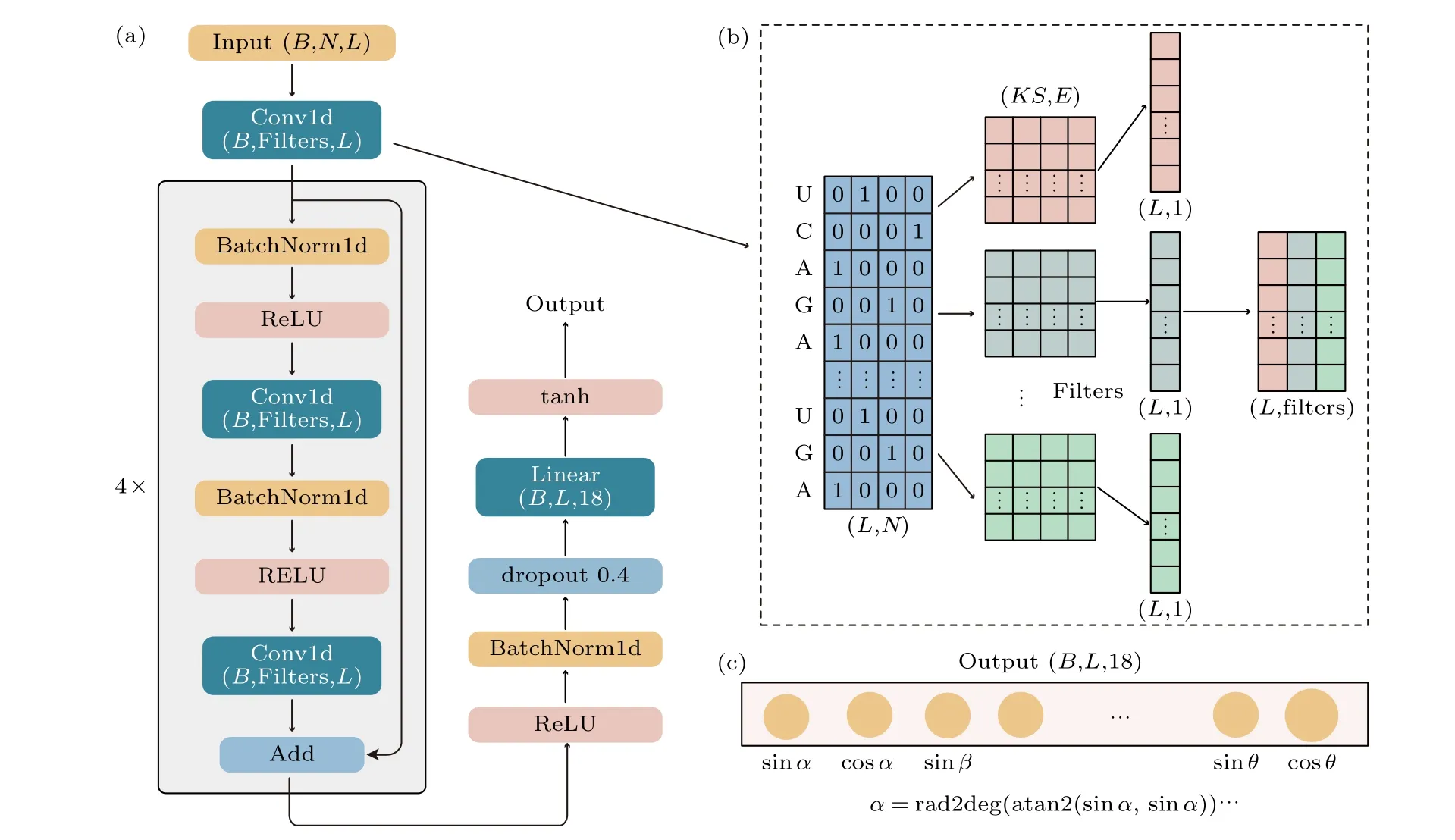
图2 DRCNN (a) 模型架构;(b) 模型中一维卷积层的原理;(c) 输出层.B,L,N,KS和Filters分别为训练中更新一次模型参数选择的序列数目、序列的长度、输入特征维度、卷积核的小大(卷积窗口一次能看到的相邻核苷酸数目)、卷积核的数目(卷积层的输出维度)Fig.2.DRCNN: (a) Network architecture;(b) Conv1d layer;(c) output layer.B,L,N,KS and Filters are batch size,sequence length,the size of the input,the size of the filter (the filter can see the number of nucleotides at one time),the number of filters.
DHLSTM模型结构如图3所示,里面的Hyper-LSTM层原理来自于文献[26],输入数据的维度是(512,8,4),模型更新一次参数选取的样本批次数目为8,描述一个核苷酸的初始特征向量维度为4;然后经过一个HyperLSTM层(这里的超参数,外部大LSTM层[27]的输出维度Hidden取64、内部小LSTM层的输出维度和改变LSTM层权重的Hypercell单元里线性投影的维度Hyper都取16;Hidden超参数64比16,32和128效果好,Hyper超参数16比32和64效果好),具体来说,第t个核苷酸特征向量和两类隐藏态进入HyperLSTM cell单元,得到第t+1个核苷酸新的特征向量和两类隐藏态,这里每个核苷酸使用不同的Hyper LSTMcell权重参数,依次算完所有核苷酸,得到描述一个批次每个核苷酸新特征数据维度(512,8,64);接着经过另一个HyperLSTM层(这里三层HyperLSTMcell单元的超参数Hidden都取64,Hyper都取16),具体来说,上一层输出的第t个核苷酸特征向量和两类隐藏态(维度(8,64))依次进入三个HyperLSTMcell单元,得到第t+1个核苷酸新的特征向量(维度(8,64))和两类隐藏态输出(维度分别为(8,64),(8,16)),依次算完所有核苷酸,得到描述一个批次每个核苷酸的新特征数据维度(512,8,64);最后将第二层HyperLSTM的输出和第一层的HyperLSTM输出相加,作为一个残差块;数据流出残差块后,进入全连接层(输入维度512,输出维度18),tanh激活函数得到输出.
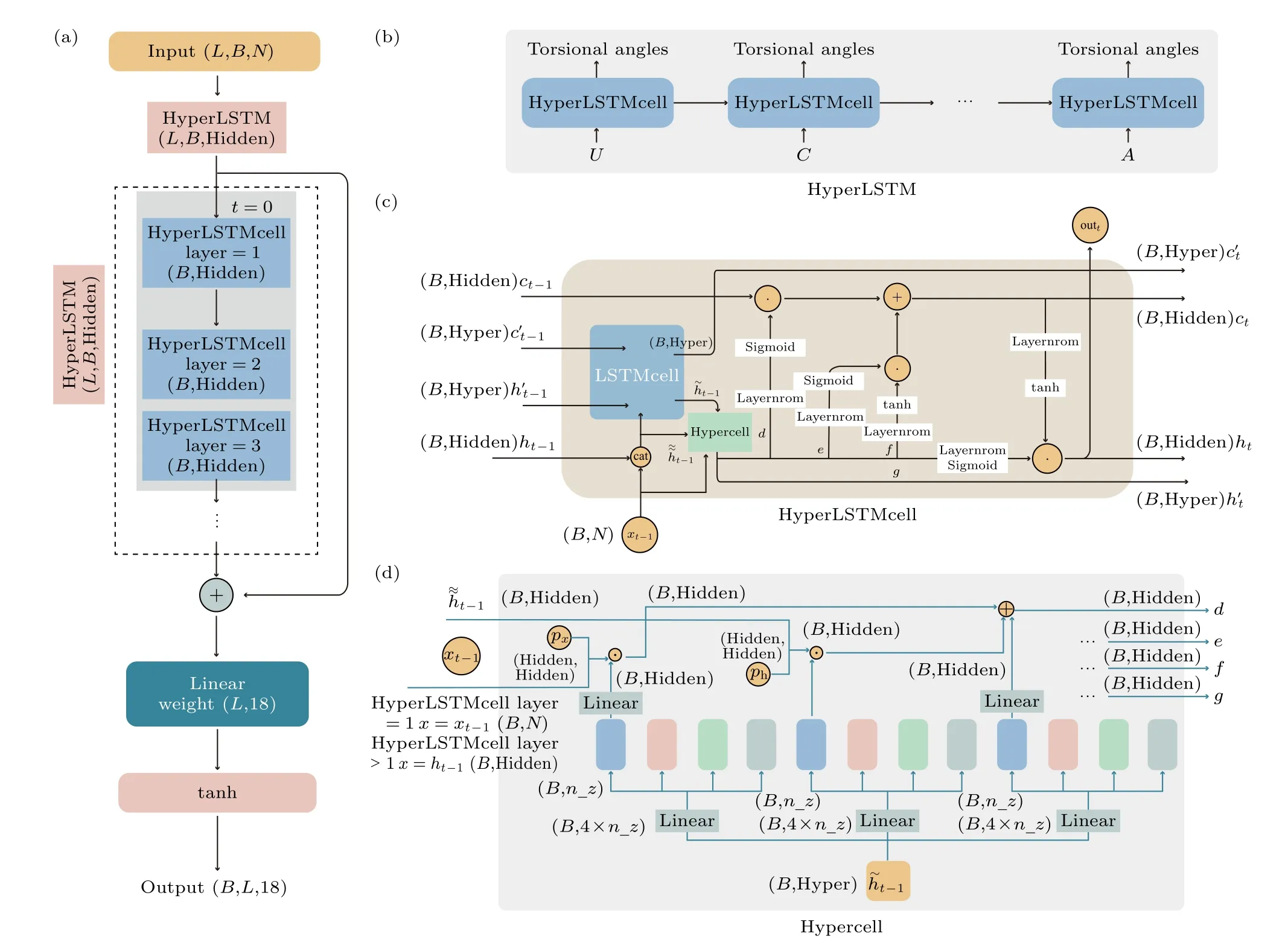
图3 DHLSTM (a) 模型架构;(b) HyperLSTM层;(c) 对每个核苷酸的处理单元HyperLSTMcell,其中ht,ct和ht -1,ct -1分别是外部更大的LSTM在t和 t -1时刻的隐藏态;,和, 分别是更小的LSTM在t和t -1时刻的隐藏态;(d) Hypercell单元.L,B,N,Hidden,Hyper和n_z分别为序列的长度、训练中更新一次模型参数选择的序列数目、输入特征维度、大LSTM层的输出维度、内部LSTM层的输出维度和改变大LSTM层权重的Hypercell单元里线性投影的维度,Px和Ph为动态可训练参数,绑定在内部超网络里,作用在输入态xt -1和隐藏态,初始值为全零张量Fig.3.DHLSTM: (a) Network architecture;(b) HyperLSTM layer;(c) HyperLSTMcell;ht,ct and ht -1,ct -1 are the states of the larger outer LSTM at time t and t -1,respectively;, and , are the states of the smaller LSTM at time t and t -1.(d) Hypercell.L,B,N,Hidden are sequence length,batch size,the size of the input,the size of the LSTM,and Hyper is the size of the smaller LSTM that alters the weights of the larger outer LSTM,n_z is the size of the feature vectors used to alter the larger LSTM weights,Px and Ph are dynamically trainable parameters,bound in the internal hypernetwork,acting on the input state xt -1 and the hidden state,and the initial value is an all-zero tensor.
DHLSTM和DRCNN训练都使用MSE损失函数和RMSprop优化器[28]训练(优化器学习率取0.001、正则化系数取0.0001,此优化器比Adam和AdamW优化器效果好,学习率0.01比0.1,0.001,0.0001和0.00001效果好,正则化系数经过尝试取学习率的百分之一0.0001比较好);同时预测9个角和单独预测一个角,预测结果基本一致,故DHLSTM和DRCNN都同时预测9个角;DHLSTM模型在训练过程中,训练损失随着epoch的增大一直下降,验证损失在第85个epoch后开始逐步上升,如图4(a)所示,故取第85个epoch的模型为最终模型;DRCNN模型在训练过程中,训练损失随着epoch的增大一直下降,验证损失在第109个epoch后开始逐步上升,如图4(b)所示,故取第109个epoch的模型为最终模型.DHLSTM和DRCNN的实现都使用Facebook的PyTorch深度学习框架[29].
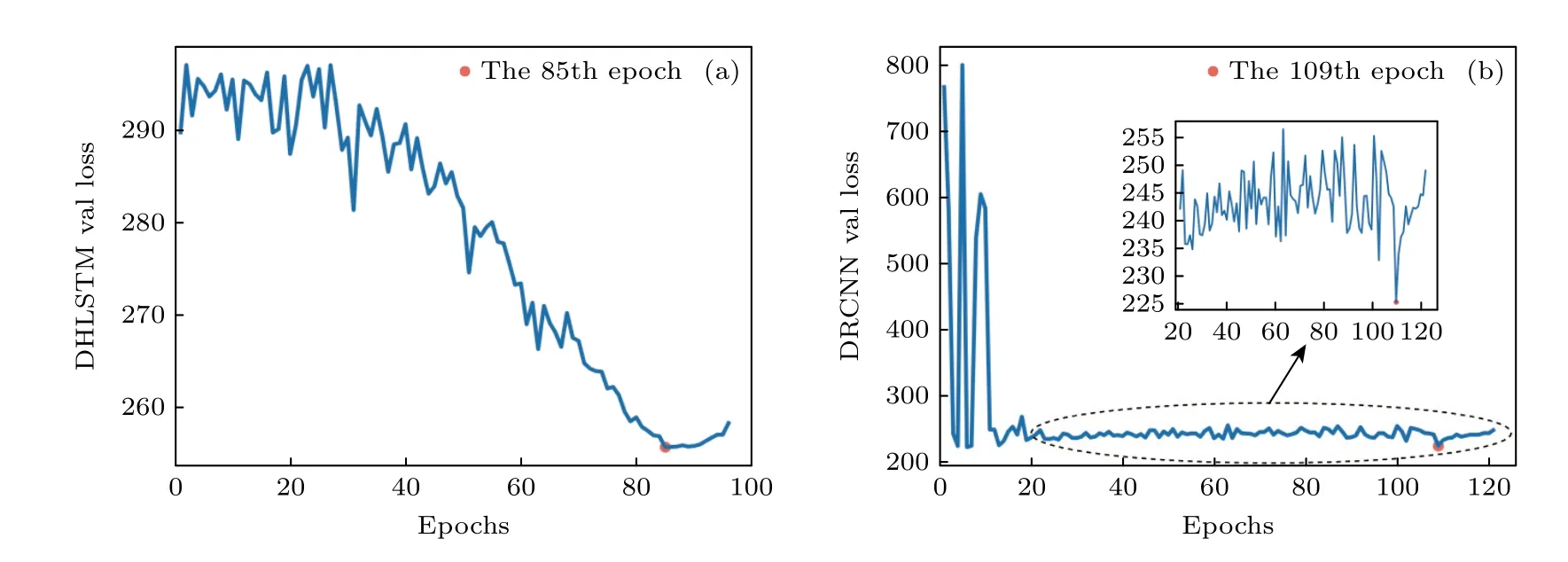
图4 (a) DHLSTM模型和 (b) DRCNN模型验证损失(MAE)随epoch的变化Fig.4.Validation loss curve with the epoch by (a) DHLSTM and (b) DRCNN.
2.2 数据集
为了比较,采用了SPOT-RNA-1D 使用的训练集、验证集和测试集(https://github.com/jaswinder singh2/SPOT-RNA-1D/tree/main/datasets)[21].训练集含有286个结构,从PDB结构数据库[30]目前可以下载到284个结构(6N5R_A,6N5L_A下架),本文训练集为这284个结构;验证集含有30个结构,都可从PDB下载;测试集有3个分别含有63,30和54个结构,从PDB数据库分别下载到62 (5Y85_B内含脱氧核苷酸下架)、30和54个结构.
SPOT-RNA-1D 数据集来自于2020年10月3日PDB数据库中所有X衍射分辨率小于3.5 Å的RNA结构;用CD-HIT-EST[31]软件对所有这些结构的序列设置相似度0.8进行聚类,多簇类中的代表序列构成训练集;然后将训练集和单簇类利用BLAST-N[32]软件设置截断值为10处理,训练集与单簇类有命中的序列被删除,单簇类中有命中的序列也被删除;经过这些处理,训练集剩下的序列作为最终训练集,单簇类剩下的序列随机分为验证集、测试集I和测试II;另外,对2021年4月5日PDB数据库中所有NMR结构,使用相同方法,去除和训练集、验证集、测试集I和测试II的冗余,作为测试集III.数据集的长度和二级结构分布信息如表1所列.
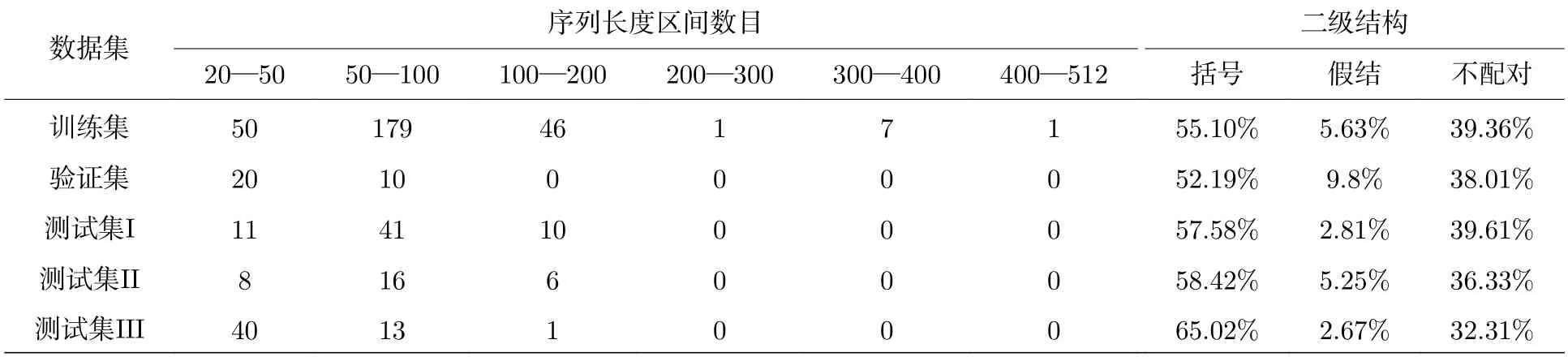
表1 训练集、验证集和3个测试集的长度和二级结构信息(百分数是数据集不同配对类型的核苷酸数目占比)Table 1.Length and secondary-structure information of training,validation and test sets.The number mentioned along with the base pairing type is the percentage of total nucleotides in the region.
2.3 输入和输出
模型的输入为核苷酸序列特征,大小为L×4的one-hot编码,四个核苷酸(A,U,G和C)分别用(1,0,0,0),(0,1,0,0),(0,0,1,0)和(0,0,0,1)表示,L为序列长度,序列长度最长为512,长度不够的补0.数据集中最长序列为414,常规做法是将所有序列用0补齐到最长序列长度.在预测时,模型预测的目标序列长度应不大于最长序列长度.这里取512是借鉴很多蛋白质模型中取值512,又观察到所有序列长度补齐到414和512的预测结果类似,故为了模型能预测更长的序列,取值512.在训练中测试过将所有序列补0区域采用mask机制,补0区域值虽然被计算但不参与下层值的计算,模型性能改善不明显.输出具体如图2(c)所示,有18个节点用于预测9个角的正弦和余弦值,然后利用atan2函数将角度的正弦和余弦值转化为角度的弧度值,再利用rad2deg函数将角度的弧度值转化为角度值.这种变换在蛋白质扭转角预测里也常用.
2.4 评 估
使用MAE评估整体性能,具体如(1)式,预测角度值和实验确定的角度值的绝对差,360°和这个绝对差的差值,取两者的小值:
3 计算结果和讨论
本文两个深度学习模型使用上面的训练集、验证集和3个独立的测试集进行训练、验证和测试.为了了解模型每个角度在每个测试集的总体表现,表2列出了DRCNN,DHLSTM和SPOT-RNA-1D[21]在验证集和3个测试集上整体的性能评估.在含有62个RNA的测试集I上,DRCNN预测的β,δ,ζ,χ,η和θ角 的MAE比SPOT-RNA-1D分别减小了5%,28%,17%,16%,24%和20%,α,γ和ε角的MAE比SPOT-RNA-1D分别增大了2%,10%和4%;DHLSTM预测的β,δ,ε,ζ,χ,η和θ角的MAE比SPOT-RNA-1D分别减小了6%,10%,9%,9%,12%,15%和11%,α和γ角的MAE比SPOT-RNA-1D分别增大了10%和13%,这表明在δ,ζ,χ,η和θ角这些角中,每层考虑相邻核苷酸特征的DRCNN比每层考虑全部核苷酸特征的DHLSTM要好,在β和ε角中,每层考虑全部核苷酸特征的DHLSTM比每层考虑相邻核苷酸特征的DRCNN要好,在α和γ角中,每层考虑间隔核苷酸的SPOT-RNA-1D比DRCNN和DHLSTM都要好.MAE值越大预测难度越大,在DRCNN中角度预测难度δ,χ,ε,β,η,θ,ζ,γ和α依次递增,在DHLSTM中角度预测难度δ,χ,β,ε,η,θ,ζ,γ和α依次递增,在SPOT-RNA-1D中角度预测难度δ,χ,ε,β,η,θ,γ,ζ和α依次递增,可以看到δ,χ,η,θ和α角在3个模型里预测难度的排序一致,考虑相邻核苷酸的DRCNN和考虑间隔核苷酸的SPOT-RNA-1D都表明ε比β容易预测,而对于DHLSTM,ε比β难预测,DRCNN和DHLSTM都表明ζ比γ容易预测,而对于SPOTRNA-1D,ζ比γ难预测.这3种方法都认为α是最难预测的,表明3个模型在角度预测难度方面有一定相似性,也各有特点.在测试集II和测试集III观察到类似的性能趋势,表明模型对不同类型的测试集具有鲁棒性.
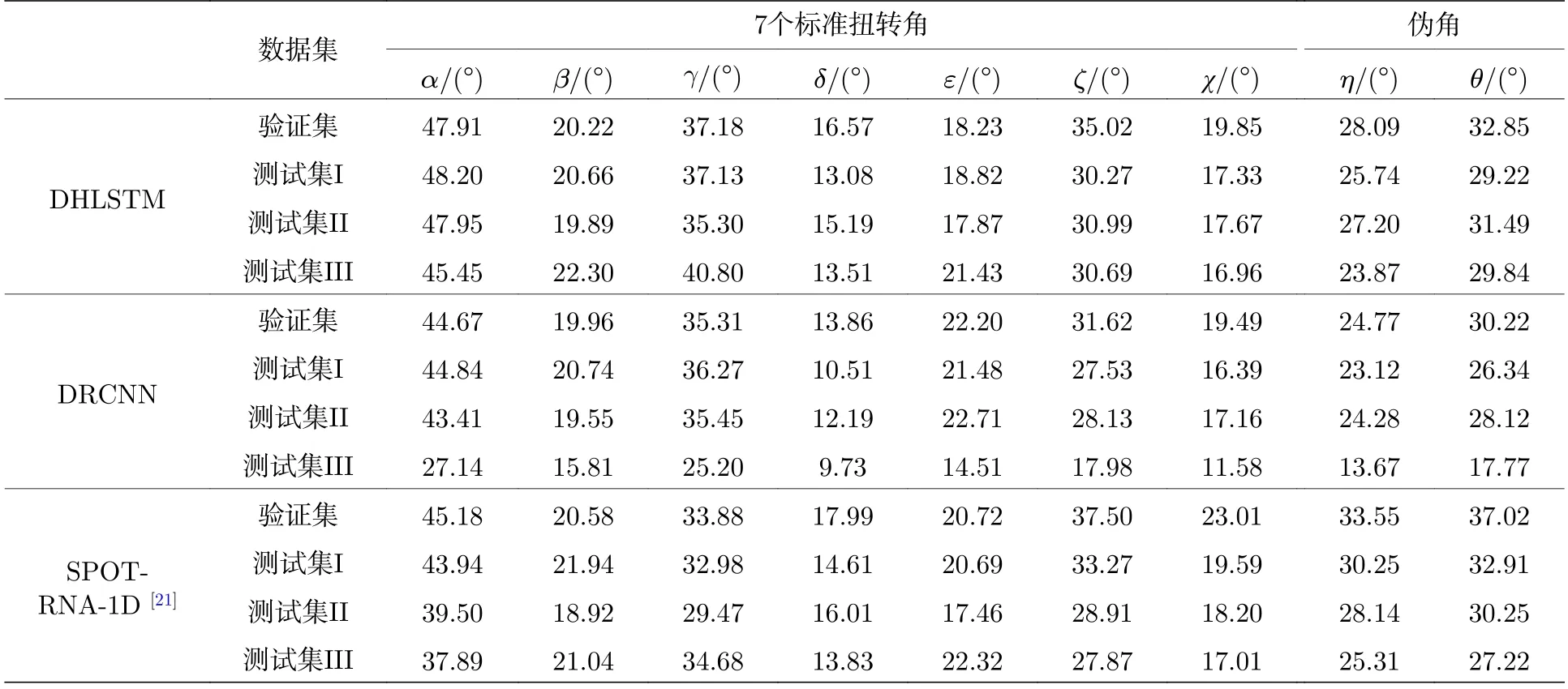
表2 DHLSTM,DRCNN和SPOT-RNA-1D在验证集和3个测试集上的MAETable 2.Performance comparison in terms of MAE on validation sets and three test sets by three models.
为了了解模型在单个序列上的表现,图5 给出了DRCNN,DHLSTM和SPOT-RNA-1D在3个测试集上单个RNA分子扭转角预测的MAE分布图,其中SPOT-RNA-1D绘制每个盒子需要五类值(最大值、最小值、中位数、上下四分位数和异常值),由论文图形数据获取工具 WebPlotDigitizer[33]得到.每个模型在3个数据集9个角度的27个MAE最小值上,DRCNN占18次,DHLSTM占3次,SPOT-RNA-1D占6次,而在27个MAE最大值上,DRCNN占4次,DHLSTM占8次,SPOTRNA-1D占15次,表明考虑相邻核苷酸特征的卷积模型DRCNN最有可能预测到最小的MAE值,DHLSTM次之,SPOT-RNA-1D很难预测相比比较小的MAE值.箱子越窄意味着每次预测MAE变化更小,模型预测更稳定,每个模型在3个测试集9个角度的27个箱子中,DRCNN出现9次,DHLSTM出现15次,SPOT-RNA-1D出现3次,表明预测最稳定的模型是考虑全部核苷酸特征的DHLSTM,且性能中规中矩,其次是DRCNN,对样本反应比较敏感的是SPOT-RNA-1D.在27个盒子相对较小的中位数上,DRCNN占18次,DHLSTM占2次,SPOT-RNA-1D占7次,表明DRCNN预测的一半数目链的总MAE比其他两个模型值要低.在异常值方面,3个测试集9个角度上,DRCNN,DHLSTM和SPOT-RNA-1D出现的异常值的数目分别为24,21和38,且DRCNN和DHLSTM出现的异常值本身是比较小,同样表明DHLSTM预测比较稳定.以上说明,考虑相邻核苷酸特征的DRCNN模型性能整体更强大,考虑全部核苷酸特征的DHLSTM模型预测更稳定.
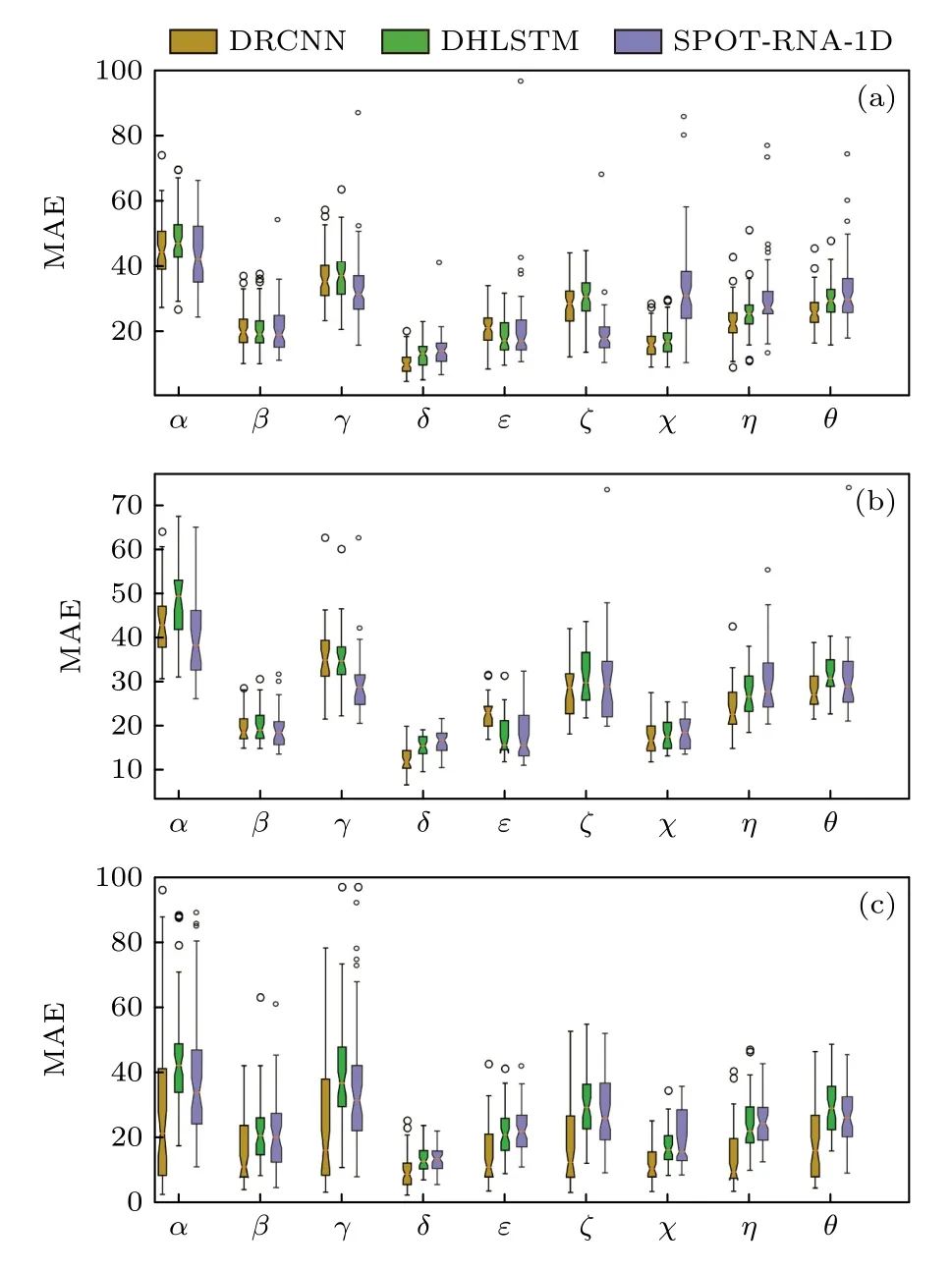
图5 DRCNN(黄色)、DHLSTM(绿色)和SPOT-RNA-1D(紫色)在测试集I (a)、测试集II (b)和测试集III (c)上单个RNA链的MAE分布图.每个盒子显示出一组数据的最大值、最小值、中位数、上下四分位数和异常值Fig.5.Distribution of MAE for individual RNA chains on test set I (a),test set II (b) and test set III (c) by DRCNN predictor (yellow),by DHLSTM (in green) and SPOTRNA-1D (in purple).Each box shows the minimum,the maximum,the sample median,the first and third quartiles and outlier.
另外绘制了角度的实验值分布,如图6橙色虚线所示,可以看出每个角度的实验值的分布是比较陡峭的,大部分角度都集中在跨度在40°左右的角度空间,有少部分角度值分布在跨度在360°的角度空间中,最容易预测的δ角跨度也是最窄的,最难预测的α角分布有3个峰,跨度是最广的.为了了解本文模型在预测分布上的能力,绘制了DRCNN和DHLSTM在测试集I的预测分布如图6黄色和绿色虚线所示,DRCNN预测所有的角度分布都比DHLSTM好;在测试集II和测试集III上,DRCNN在β和γ角上预测的分布比DHLSTM要好,两个模型在预测其他7个角的分布类似.
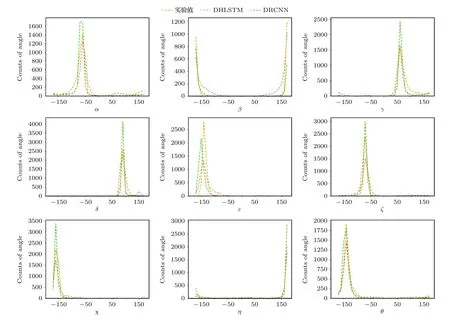
图6 测试集I扭转角的实验值(橙色)、DHLSTM预测值(黄色)和DRCNN预测值(绿色)分布图Fig.6.Distribution plots of native (in orange),DHLSTM predicted (in yellow),and DRCNN predicted (in green) nine torsion angles on test set I.
二级结构对RNA建模起着重要角色,根据DSSR软件[34]输出的RNA二级结构,可将RNA二级结构分为三种类型,括号(['(',')']),假结(['[',']','{','}','<','>','A','a']),环 区['.'].比较了DRCNN和DHLSTM在测试集III中对3种二级结构类型的整体预测性能(表3),可以看出,对DRCNN和DHLSTM来说括号类型的核苷酸的扭转角最容易预测的,处于环区的核苷酸的扭转角是最难预测;还可以观察到,DRCNN预测3种类型的MAE误差都比相应的DHLSTM预测的要低;在其他两个测试集观察到同样结果,因此,扭转角预测的误差主要来自于环区和假结区域,在预测括号、假结和环区区域的扭转角上DRCNN都比DHLSTM好.
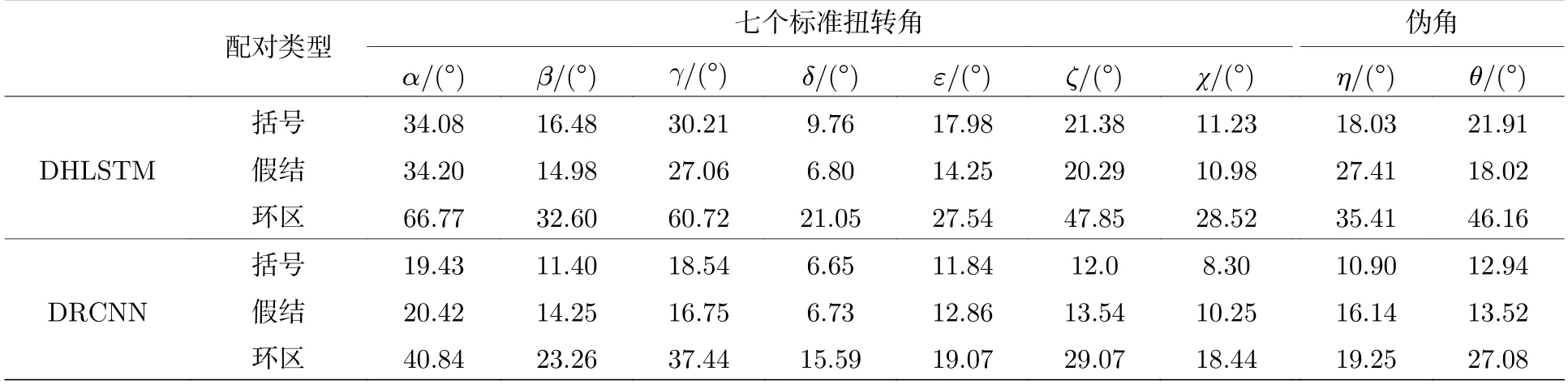
表3 DHLSTM和DRCNN在测试集III不同配对类型中扭转角预测的MAETable 3.Performance according to mean absolute error by DHLSTM and DRCNN for nucleotides in different pairing type on test set III.
表1统计了训练集、验证集和3个测试集的序列长度分布.由表1可以看出,在训练集和验证集中各个长度分布并不均匀,长度在50到100区间的有179个结构,在100到200区间的只有46个.为了了解这种差异是否会导致DRCNN和DHLSTM对长RNA扭转角预测性能较差,图7绘制了两个模型在9个角度上的表现与序列长度的关系.观察DHLSTM和DRCNN的预测结果,9个角的MAE值在数目少的长度区间[78,94],[155,171]和[171,186]并不大;还观察到DRCNN在短长度区间[1,47]结果比DHLSTM结果好;因此,虽然训练集和验证集对不同长度的RNA数目分布不均匀,但并没有造成DRCNN和DHLSTM在预测上的长度偏好.
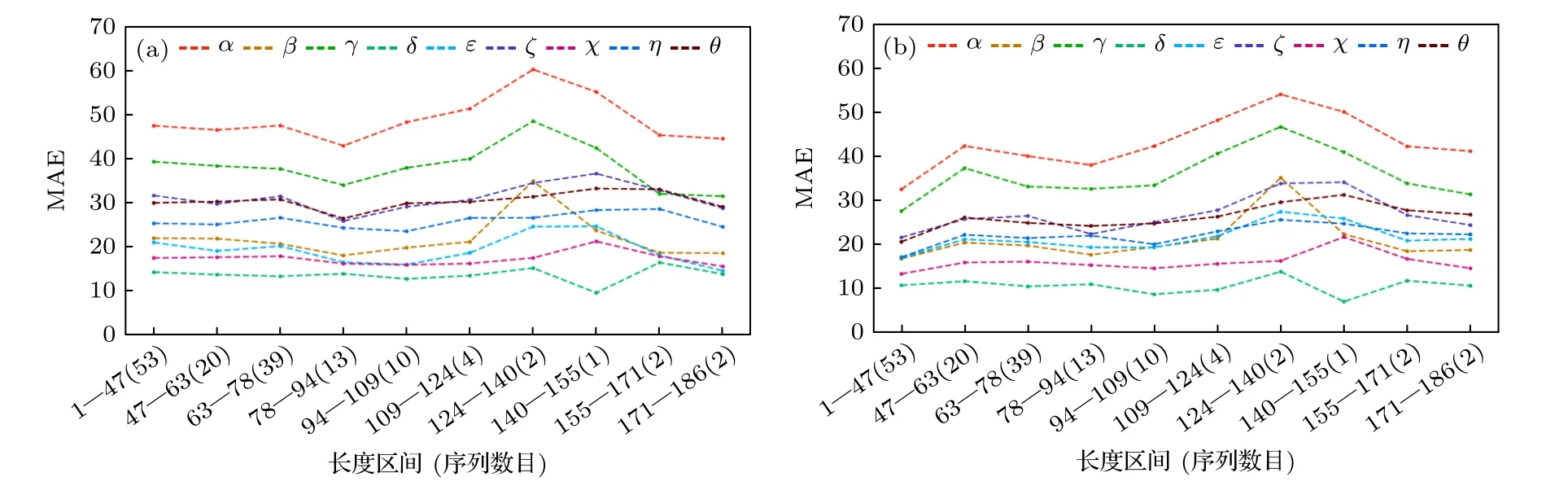
图7 (a) DHLSTM和 (b) DRCNN分别在3个测试集(147个RNA)的9个扭转角的MAE与RNA序列长度的函数Fig.7.On 147 RNAs in the three test sets,the MAE is measured as a function of the length for the nine torsion angles by (a) DHLSTM and (b) DRCNN.
和SPOT-RNA-1D方法一样,为了了解扭转角之间的相关性,在测试集I上绘制了如图8所示的扭转角相关矩阵.一般情况下,相邻扭转角之间高度相关,而较远扭转角相关性较小,但是矩阵显示,对于DRCNN和DHLSTM,α和γ角有很强的相关性,两者也是模型预测难度最大的两个角,ζ和θ有最强的相关性,两者预测难度排名也是相邻的.在其他两个测试集的结果相同.
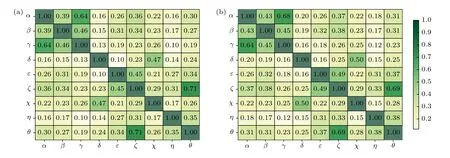
图8 (a) DHLSTM和 (b) DRCNN分别在测试集I上扭转角的MAE的相关系数(CCs),值越大表示两个角度越相关Fig.8.Correlation coefficient (CCs) for MAE of between the nine torsion angles of test set I by (a) DHLSTM and (b) DRCNN,the larger the CC value,the more correlated between the two torsions.
观察一条链中预测的每个角度,预测的大部分扭转角比一些近天然态或者类天然态结构的扭转角更接近天然态结构扭转角的值.和SPOT-RNA-1D方法一样,也测试了DRCNN和DHLSTM这两种深度学习模型预测的角度和不同RMSD结构的角度之间的差异是否可以用于结构的质量评估.为此,使用3dRNA[3]测试集85个RNA和它们的decoys进行了测试.图9绘制了DRCNN和DHLSTM在其中一个RNA(PDB ID号1Y69,链9)在预测角度与诱饵模型结构角度之间的MAE和结构精度的函数关系,MEA随RMSD持续增加.在85个数据集中的其余84个RNA中也观察到类似的趋势,这表明与模型预测角度的偏差或结合其他参量可用于模型质量评估.
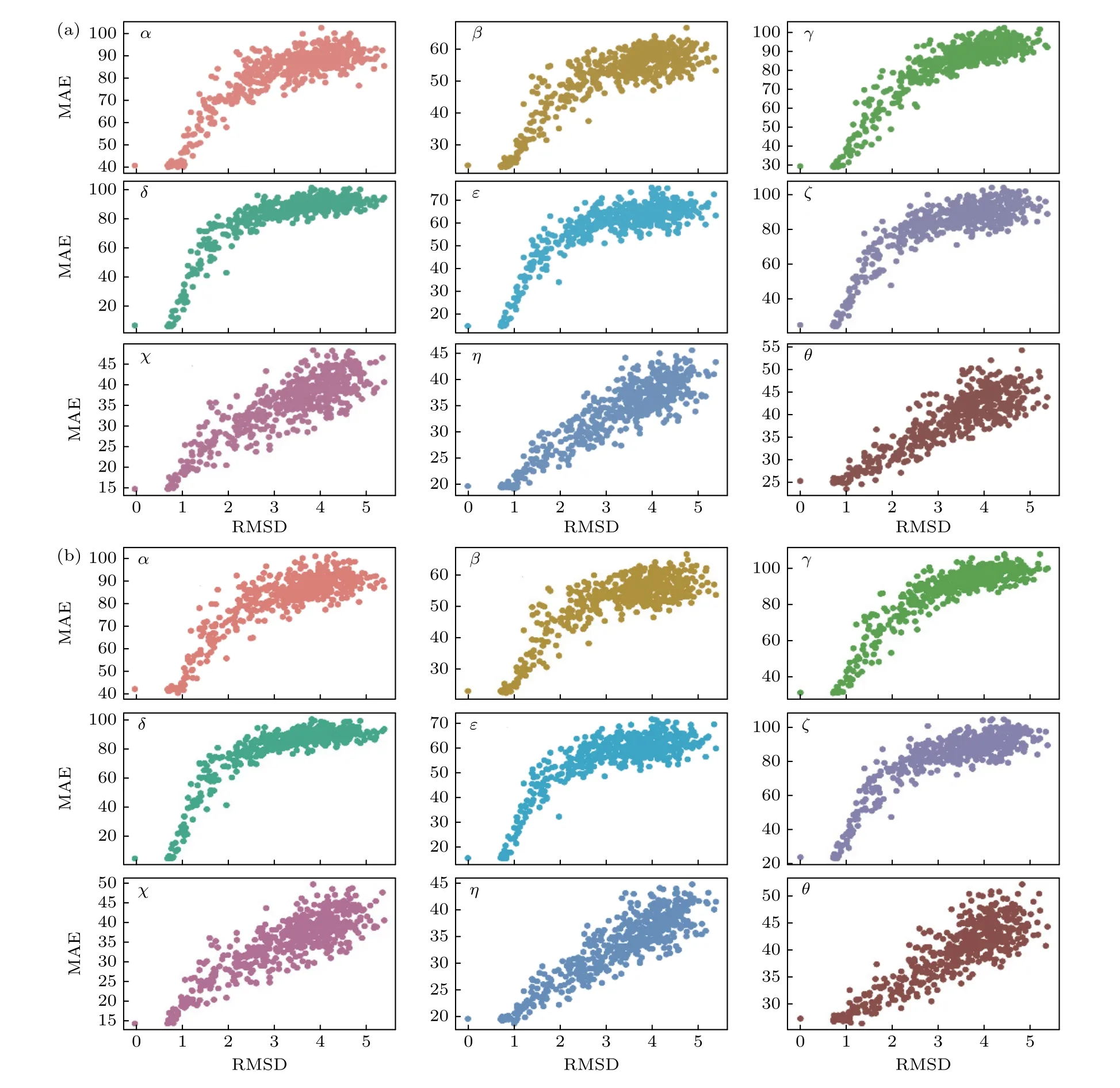
图9 (a) DRCNN和(b) DHLSTM分别在RNA 1Y69(链9)上预测角度与decoys结构角度之间的MAE与RMSD的关系Fig.9.On RNA 1Y69 (chain 9),the MAE is measured as a function of RMSD for the nine torsion angles by (a) DRCNN and(b) DHLSTM.
4 结论
本文提出了一种预测RNA分子扭转角的深度学习方法1dRNA,采用了DRCNN和DHLSTM两个基于时序网络的模型去预测RNA的7个扭转角(α,β,γ,δ,ε,ζ和χ)和2个伪角(η和θ),并和现有方法SPOT-RNA-1D进行了比较.结果表明不同网络在不同角度上各有优势,当序列长度不超过50时,在预测9个角时,考虑相邻核苷酸特征的DRCNN比考虑全部核苷酸特征的DHLSTM和考虑间隔核苷酸特征的SPOT-RNA-1D都好;当序列长度超过50,在δ,ζ,χ,η和θ角这些角中,DRCNN预测的结果整体上比DHLSTM和SPOTRNA-1D要好,在β和ε角中,DHLSTM预测的结果整体上比DRCNN和SPOT-RNA-1D要好,在α和γ角中,SPOT-RNA-1D预测的结果整体上比DHLSTM和DRCNN要好;3个模型在9个角度的预测难度上类似,角度的实验值和预测值分布可以看出角度预测的难度主要在于角度分布的复杂程度,分布越复杂越难预测,DRCNN和SPOTRNA-1D预测出来的角度分布比DHLSTM丰富;序列环区的角度分布比配对区域复杂,角度预测难度也比配对区域大很多;每个模型在链长度集中在非长链区的训练集和验证集上训练,但在预测时对长链预测效果也不错;在模型预测稳定性上,考虑全链核苷酸的DHLSTM比考虑相邻核苷酸的DRCNN和考虑间隔核苷酸的SPOT-RNA-1D要稳定很多,异常值少;模型的各个结果在3个测试集上表现类似,表明模型性能对不同数据集稳定.从结果来看,面对比较短序列,9个角度都用考虑相邻核苷酸特征的卷积网络更好,当序列长时,在预测δ,ζ,χ,η和θ角用考虑相邻核苷酸特征的卷积网络更好,预测β和ε用考虑全链核苷酸特征的超循环网络更好,预测α和γ角用考虑间隔核苷酸特征的膨胀卷积网络更好.在数据集方面,尝试过加入新发表的RNA结构增大数据集训练,精度能提高但不明显;可以设计其他类型的网络,尝试使用单纯的全连接网络和Transformer[35]网络训练,角度预测整体MAE比DRCNN和DHLSTM更好,但预测的角度分布很差,很难预测出角度分布峰值之外的区域;尝试过在DRCNN和DHLSTM这个两个模型上改进,精度能提高但不明显;在加入新特征方面,加入二级结构特征,能提高精度但也不明显.在改进角度预测方面,从结果可以看出角度分布决定了预测难度,在预测前如何预先处理这种分布,和如何把这种分布加入损失函数,应该可以很大提高预测精度;另外直接预测角度实值难度大,可以考虑将跨度360°的角度分布分成36个bin去预测.
