二氧化碳电催化剂理论设计方法研究进展*
2024-01-06王清华肖一杨杨应举白红存
王清华,肖一杨,杨应举,,刘 晶,白红存
二氧化碳电催化剂理论设计方法研究进展*
王清华1,肖一杨2,杨应举2,†,刘 晶2,白红存3
(1. 国家能源集团合肥发电有限公司,合肥 230026;2. 华中科技大学 能源与动力工程学院,煤燃烧与低碳利用全国重点实验室,武汉 430074;3. 宁夏大学 化学化工学院,省部共建煤炭高效利用与绿色化工国家重点实验室,银川 750021)
CO2电催化还原(ECR)是一种极具应用前景的CO2利用技术,其关键在于高性能催化剂的开发。采用理论方法可有效指导与加速高效ECR催化剂的设计。从密度泛函理论(DFT)、溶剂化模型、电化学计算模型和机器学习四个方面介绍了ECR催化剂的理论设计方法。DFT、DFT + U、杂化泛函可有效计算ECR反应体系的能量、电子特性等,预测催化剂的性能;对于ECR反应中的溶剂效应,综合计算成本和精度需考虑显式溶剂化模型、隐式溶剂化模型和混合模型;在ECR电化学计算中,恒定电极电位模型比计算氢电极模型更能有效描述CO2还原的能量变化;机器学习可高效、低成本地实现ECR催化剂的性能预测、活性位点设计和组分优化。最后,对CO2电催化剂的理论设计方法进行了展望。
CO2电催化还原;密度泛函理论;电化学计算;机器学习
0 引 言
化石燃料的过度利用造成了大量的CO2排放,导致温室效应、海洋酸化、海平面上升等环境问题[1]。因此,减少CO2排放具有至关重要的意义。利用可再生电力将CO2电催化还原(electrocatalytic reduction, ECR)为CO、HCOOH、CH4、C2H4等工业原料,实现CO2储存和清洁能源转换的双重目的,近年来受到工业界和学术界的极大关注[2]。ECR技术具有反应条件温和、反应物廉价、工艺绿色、高效率等独特优势和广阔的应用前景[2]。
CO2作为一种完全氧化的线型分子,其标准生成焓为 −393.5 kJ/mol,通常情况下热力学稳定性较强,为CO2电催化还原过程带来了巨大的挑战[2]。在典型的CO2电解槽中,阳极和阴极由离子交换膜隔开。在阳极,水被氧化成分子氧;在阴极,CO2发生电化学还原反应。由于CO2还原路径的复杂性与还原电位的相似性,使得ECR选择性地生产理想的化学品具有较大的挑战性[3]。然而,在基于水电解质的CO2电解槽中,电解池阴极侧的CO2电催化还原会与析氢反应(hydrogen evolution reaction, HER)竞争。因此,ECR技术工业化应用的关键在于设计并开发高效的催化剂,高效获得目标产物的同时,抑制析氢副反应的发生。目前,大量催化剂的ECR性能已被研究,包括金属催化剂、金属硫化物催化剂、单原子催化剂、非金属催化剂等[2]。然而,现有催化剂的性能难以满足ECR工业化应用的需求。
潜在的催化剂材料种类较多,相较之下,实际合成的材料却相当有限,因此采用理论方法,通过催化剂的组成、结构和性能之间的关系指导与加速先进ECR催化剂的设计具有非常重要的意义。例如,密度泛函理论(density functional theory, DFT)和动力学可以在原子尺度上直接研究机理,识别描述符和活性位点[2]。在此基础上,可以进一步指导材料设计合成,并对未知材料的催化性能进行预测。此外,随着机器学习算法的发展以及DFT数据库的建立,高通量筛选方法在ECR催化剂合成中也起到愈发重要的作用[2]。
本文综述ECR催化剂设计中常用的理论方法,包括密度泛函理论、溶剂化模型、电化学计算模型和机器学习,对比各种方法的优势和劣势。讨论不同计算方法、模型的应用,最终提出ECR催化剂理论设计方法的未来展望。
1 密度泛函理论
1.1 密度泛函理论简介
密度泛函理论是描述分子和材料的电子结构、热力学和动力学性质的强有力工具,对催化计算化学的进步做出了重大贡献[2]。目前的DFT方法本质上是求解薛定谔方程的近似方法,也是最为广泛采用的一种。
目前,DFT已经发展出三条基本定理,即Hohenberg-Kohn(HK)定理I、HK定理II以及Kohn-Sham(KS)理论。Hohenberg和Kohn于1964年首先提出并证明了DFT的两个基本定理,证明了基态密度和外部势之间的一一映射关系(HK定理I);随后提出的第二个定理(HK定理II)表明,使总能量最小的密度(电子密度的函数)是系统的精确基态密度。然而,Hohenberg和Kohn的理论并不包含任何关于能量密度泛函的具体表达式。因此,必须结合近似动力学和交换相关泛函(exchange-correlation functional, ECF)将DFT付诸实践。KS理论[4]将相互作用的多体问题映射到非相互作用的单粒子问题上,其中一组非相互作用轨道(KS轨道)被一致地求解,总密度通过已占据的非相互作用轨道密度求和来计算,在非相互作用轨道上应用单粒子动能算符来近似计算动能,该理论显著提高了DFT实际应用中动能贡献的准确性。KS-DFT的整体准确性取决于交换相关泛函的公式,其必须考虑到相互作用和非相互作用电子之间的动能差异,以及准确捕获交换和相关的量子效应。
交换相关泛函常用的两种形式是局部密度近似(local density approximation, LDA)[5]和广义梯度近似(generalized gradient approximation, GGA)[2],其中Perdew-Burke-Ernzerhof(PBE)泛函是应用最为广泛的泛函之一。随着计算能力的发展,使用LDA和GGA的DFT计算可以在合理的计算时间内研究包含数百个原子的表面电化学系统。对于金属体系,DFT计算的结果通常是可靠的。
1.2 DFT + U和混合DFT
采用LDA和GGA的DFT计算在描述过渡金属氧化物、稀土元素等物理性质时难以得到准确结果,该方法无法准确考虑过渡金属d或f电子的强电子相关性。但电催化常用的一类材料是过渡金属氧化物、过渡金属硫化物等。因此,需要超越传统的DFT方法,纠正这些强相关电子的自洽场误差(self-interaction error, SIE)和过度离域。两种最常见的理论是DFT + U和杂化泛函。
DFT + U最初是由ANISIMOV等[6]提出的,其核心思想是在哈特里−福克(Hartree-Fock)理论中专门处理强相关d电子的交换相关泛函相互作用,使用传统DFT方法处理系统的其余部分。原子内Hartree-Fock处理用于去除局域d电子的自洽场误差。DFT + U方法需要选择一个U值(U值是密度泛函理论计算中使用LDA + U方法考虑相关效应的一个参数),实际上是DUDAREV等[7]提出的U/J,U/J对应于原子内库仑/交换相互作用。通常,不同U值的理论计算结果与已知材料的实验观测值进行比较,从而选择理论计算结果与实验结果接近的U值。在这种情况下,DFT + U计算必须考虑半经验而不是完全从头算。目前已经提出了几种确定U值的第一性原理方案,例如确定U的约束DFT计算[8]等。DFT + U的主要缺陷是选择一个U值,且U值的选择具有非唯一性,这部分降低了DFT + U的可靠性。DFT + U的优点在于计算成本仅略高于标准DFT,这使其成为过渡金属氧化物、硫化物等电化学系统的实用方法。另一种自洽场误差校正方法是杂化泛函。其核心思想是将精确Hartree-Fock交换的一小部分包含到应用于系统中所有电子的交换相关泛函中。常用的杂化泛函,例如B3LYP泛函[9],其含有20%的Hartree-Fock交换,是研究分子的常用泛函。杂化泛函需要对非局部Hartree-Fock交换进行充分的计算,计算成本很高,特别是对于周期系统。因此,杂化泛函方法的应用比DFT + U方法更有局限性。
综上所述,以LDA或GGA作为交换相关泛函的DFT计算效率最高。DFT对结构优化通常是可靠的;弛豫的结构可以作为输入,用于更高精度、更昂贵的其他性能计算。对于金属体系等,标准DFT通常也可用于计算电子结构(例如态密度、原子电荷等)和各种热力学性质(能量、状态方程等)。对于含有过渡金属离子的材料(例如过渡金属氧化物等强相关电子材料),需要采用DFT + U方法,甚至更昂贵的杂化泛函方法。
1.3 基于DFT的机理研究与催化剂筛选
DFT常与微观反应动力学相结合,预测催化剂的性能或探究催化剂性能强化的机理[10]。例如,ZHONG等[11]采用DFT计算了CO在金属催化剂表面的吸附能,结合微观反应动力学探究了CO吸附能与催化剂活性和选择性的关系,并确定了CO吸附能的最佳区间。此外,计算了中间体的吸附能,进而探究反应的活化能垒。结果表明,CuAl表面上C2H4形成的能垒低于Cu表面,表明CuAl双金属催化剂比Cu具有更高的C2H4法拉第效率。同时,通过高通量计算,筛选出有利于C2H4生成的活性位点,并探究了CuAl合金的最佳活性位点。
2 溶剂的影响
CO2电催化还原中,电极与电解质溶液直接接触,其中水、乙腈和二甲基甲酰胺是使用最广泛的极性溶剂。为了模拟CO2电催化还原实验的真实情况,考虑溶剂效应非常重要。部分常用的溶剂化模型总结如下。
2.1 显式溶剂化模型
显式溶剂化模型,即在电子结构计算中直接包含溶剂分子。溶剂、溶质、吸附剂和电极表面之间的相互作用可以通过该模型精确地描述。然而,显式溶剂化模型的计算成本过高。量子力学/分子力学方法(quantum mechanics/molecular mechanics, QM/MM)用量子力学只描述溶质附近的少数溶剂分子,而其余的溶剂分子则用经典的分子力学方法处理,该方法有效减少了采样时的计算成本[12]。对于非均相电化学反应,电极表面和吸附物需要进行量子力学处理,而溶剂区域可以由经典力场控制的显式溶剂分子进行建模。然而,QM/MM方法也带来如何处理QM和MM区域之间的边界概念问题[13]。
2.2 隐式溶剂化模型
隐式溶剂化模型采用“反应场”的概念表示,该模型的溶剂被认为是具有介电常数ε的均匀介质[14],如图1所示。溶剂中的电荷分布表示为电场,该电场因溶质的存在而极化并对其作出响应。隐式溶剂化模型与显式溶剂化模型的区别在于大量的单个溶剂分子被移除,转而被具有与溶剂一致性质的连续介质填充。两种常用的隐式溶剂化模型是类导体溶液模型[15]和溶质电子密度的溶剂化模型[16]。类导体溶液模型将连续介质视为导体,极大简化了溶质极化连续介质引起的静电稳定相互作用的计算。溶质电子密度的溶剂化模型基于广义玻恩近似,其近似泊松方程和泊松−玻尔兹曼方程,因此可以解析求解。隐式溶剂化模型比显式溶剂化模型节约计算时间和成本,但是不能描述溶质和溶剂分子之间的特定相互作用,例如氢键。
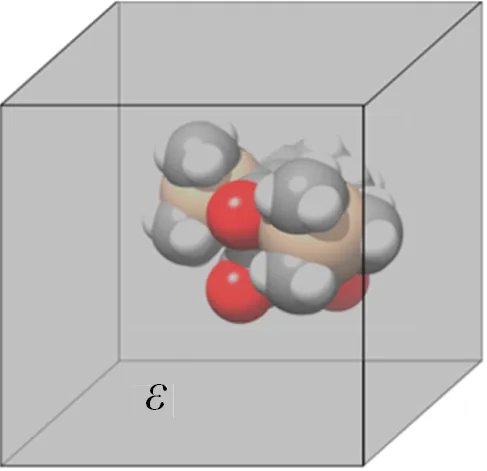
图1 介电常数为ε的连续介质模拟隐式溶剂化模型[14]
2.3 混合隐式−显式溶剂化模型
隐式和显式溶剂化模型相结合,出现了混合隐式−显式溶剂化模型,可平衡准确性和计算效率。混合溶剂化模型用溶剂分子的原子级表示来处理靠近溶质/电极的溶剂区域,而较长范围的剩余溶剂区域则由隐式溶剂化模型处理,与溶质相关的氢键被明确地解释,同时仍然考虑到较长范围的静电响应。例如,在GaP表面上的CO2光电还原研究中[17],GaP表面采用显式溶剂化模型处理,而其余区域则由隐式溶剂化模型处理。
此外,LESSIO等[18]采用混合隐式−显式溶剂化模型计算了GaP(110) 电极表面上吡啶催化CO2还原的反应动力学能垒,研究了CO2还原反应中间产物的形成过程,如图2所示。其中,反应步骤E(PyH*sol+ H−* →-DHP*)具有更低的动力学能垒,约为104.5 kJ/mol,从热力学的角度上说明更利于CO2还原反应的进行。

图2 吡啶催化CO2还原过程中氢转移反应的二维势能面[18]
3 电化学计算模型
3.1 计算氢电极模型
计算氢电极(computational hydrogen electrode, CHE)模型是一个简单的模型,将DFT计算涉及的质子耦合电子转移(proton-coupled electron transfer, PCET)的能量转换为应用电位下的能量[19]。转换能量通常参考实验中常用的两种参比电极:可逆氢电极(reversible hydrogen electrode, RHE)和标准氢电极(standard hydrogen electrode, SHE)。对于RHE模型,当一对质子和电子与分压力为1 × 105Pa的气体H2处于平衡状态时,定义为零电压:
H++ e−↔ 1/2H2,= 0 V (vs. RHE) (1)
式中:为施加的电势。当质子和电子与气态H2处于平衡状态时,质子和电子在0 V下相对于RHE的化学势是气态H2在任何pH和温度下减去 −化学势的一半,其中表示转移的电子数。
对于SHE模型,质子浓度对质子和电子的化学势的影响如下:
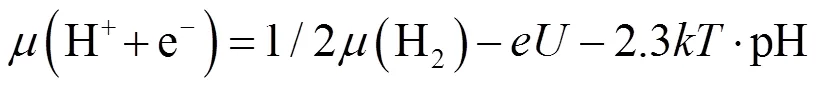
式中:、和分别为化学势、玻尔兹曼常数和温度。CHE模型允许计算基元反应的自由能变化,不需要比非电化学条件下更多的计算。
过电位是进行电化学反应时,除了平衡电位外施加的额外电位,是评价催化活性的关键指标之一,这里的平衡电位是总的催化反应循环能量除以转移电子数。为了得到CHE模型下的过电位,需要引入电势决速步骤(potential-determining step, PDS)和极限电势(L)的概念。其中,PDS表示质子耦合电子转移步骤中自由能最高的步骤,PDS决定了催化反应循环的整体过电位。L定义为PDS处自由能变化的负值,表示还原反应的最大电极电势,计算公式如下:
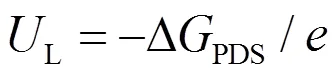
因此,CHE模型得到的热力学过电位()由L与平衡电位(eq)之差的绝对值定义:
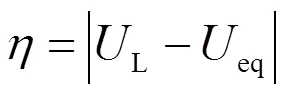
尽管CHE模型使用起来很简单,但存在一些缺陷:
(1)CHE模型不能应用于过渡态,因此CHE模型得到的PDS不能代表实验测量获得的决速步(rate-determining step, RDS)。
(2)大多数DFT计算是中性条件下进行的,在计算过程中,电极电位随系统中的溶剂而变化。因此,反应物状态、过渡状态和生成物状态的电极电位有所不同[20]。在实际条件下,电极电位是固定的,因此CHE模型在计算相对能量时会存在误差。
3.2 恒定电极电位模型
虽然CHE模型使用简单,但CHE模型忽略了上述电极电位变化导致的反应能和活化能误差。恒定电极电位(constant electrode potential, CEP)模型通过改变单元中电子数迭代进行DFT计算,将与目标电位匹配。参考SHE的电极电位可以用DFT计算:

式中:为费米能;SHE为SHE的绝对电位,其值为4.44 ± 0.02 V。
CEP模型通过改变单元电子数来调控费米能级(Fermi level,f)。然而,这会造成净电荷的不平衡,因此需要反电荷[21]。通常通过使用隐式溶剂和线性化泊松−玻尔兹曼方程来放置连续反电荷,其中反电荷由德拜长度决定[21]。目标电位下,带电单元电解池的总电子能量()可以通过以下公式进行计算:

式中:DFT为通过DFT计算得到的带电单元的电子能;e为电解池中的电子数;e为电子的化学势,等于参考体电解质区域静电势的费米能;ee表示电子移除或添加的能量。
CHE模型仅适用于质子耦合电子转移步骤,而CEP模型可以研究电极电位对不涉及质子转移的反应的影响,例如电化学CO2还原过程中CO2自由基阴离子在金属表面的吸附。GARZA等[22]发现施加的电势改变了CO2吸附能,进而改变了CO2还原产物的种类,如图3所示。
3.3 从头算分子动力学
从头算分子动力学(ab initio molecular dynamics, AIMD)是一种加速采样方法,用于探索系统的自由能。在分子动力学模拟过程中,可以通过在系统的真实能量场中添加正高斯势来加速计算。如果模拟时间足够长,则可以从所有附加偏置势的总和中获得多维自由能图。其缺点是必须选择一个循环伏安曲线,这可能对结果有所影响。高斯势函数的选择也可能影响结果,尽管预计影响较小。
ZHAO等[23]采用从头算分子动力学和缓慢增长取样方法计算了Ni单原子掺杂石墨烯对CO2电催化还原合成CO的反应动力学能垒,如图4所示。发现活性位点的电荷存储能力和吸附中间体的氢键对催化剂性能具有显著的影响,然而该影响却被密度泛函理论计算忽略了。储存高电荷容量的活性位点可以为电化学反应提供电子,降低反应的动力学能垒。通过稳定反应中间体并促进质子转移,氢键促进极化反应中间体的形成,进而促进CO2电催化还原反应。
4 机器学习
4.1 机器学习简介
机器学习(machine learning, ML)是驱动计算机智能化的基础,可以使机器具备自动学习的能力,实现非显式编程。目前,机器学习功能强大,可以快速提取隐藏在大量数据中的有用信息,在各个领域都有重要的应用。机器学习可以通过拟合与潜在催化剂性质相关的输入和输出数据中的复杂函数来构建非线性映射,与使用量子化学计算的传统计算技术相比,机器学习可以以非常低的计算成本快速预测催化性能,筛选高效的催化剂[24],如图5所示。催化剂的机器学习筛选过程包括以下步骤:(1)催化剂材料结构设计;(2)性能预测与研究;(3)催化剂材料合成。其中,材料结构设计可以枚举所有可能的催化剂,性能预测与研究可以筛选出比较有前景的催化剂材料,为下一步催化剂合成提供配方。
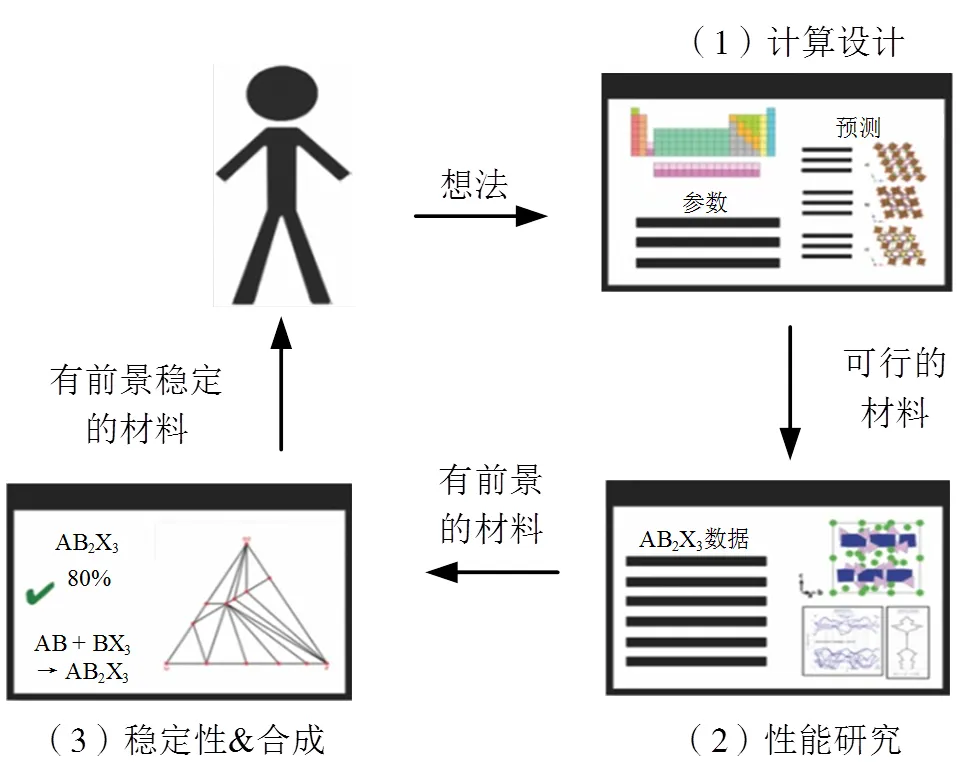
图5 机器学习在新材料设计中的应用[24]
4.2 机器学习流程
典型的机器学习方法包括数据收集、特征工程、算法选择和模型验证四个步骤,如图6所示。首先,机器学习的特点即可以从数据集中学习并分析支配数据集的潜在规则。因此,对于机器学习来说,充足且高质量的数据非常重要。特征工程包含特征提取和特征选择,是提取输入数据和训练机器学习模型的关键过程,优秀的特征工程可以确保后续构建的机器学习模型具有较高的泛用性和精确度。随后,应用适当的机器学习算法构建模型,并从给定的数据集中学习,以预测反应机制或属性。最后,对机器学习模型进行验证,然后基于训练集和测试集进行优化,最终得到具备优秀泛用性和准确度的模型。

图6 机器学习流程
在数据收集过程中,收集的数据被用来训练模型算法,以定量预测指定的属性,如CO和H吸附能等,最终目的在于寻找具有高活性、高选择性、高稳定性和低成本的催化剂。用于机器学习训练的数据可自主计算,也可通过数据库收集。材料计算学数据库的建立与积累为机器学习的应用带来了极大便利。无机晶体结构数据库(Inorganic Crystal Structure Database, ICSD)包含超过21万个晶体结构,是目前世界上最大的晶体结构数据库,广泛应用于材料科学[25]。晶体学开放数据库(The Crystallography Open Database, COD)汇集了绝大多数可用的无机、金属有机和有机分子结构数据[26]。开放量子材料数据库(Open Quantum Materials Database, OQMD)包含超过20万个DFT计算晶体结构[27]。快速访问数据库可实现数据的高效收集,为机器学习的应用提供了极大便利。然而,目前数据收集过程很大程度上仍需研究人员自主计算或实验得到,这个过程会花费大量时间。
数据收集完成后,需要采用特征工程将原始数据转化为机器学习算法所能识别的参数。因此,特征工程是为目标值选择独特相关输入参数(这些相关参数又被称为特征)的过程。特征首先需要能够被机器学习算法所识别,因此特征的形式通常是数字、向量、矩阵等。特征的选取应该是通用且有效的。在ECR催化剂筛选中,用于描述表面微观结构的常用特征有泡利电负性、原子序数、反应中间体吸附能[28]、活性位点配位数[29]等。
机器学习算法基于数据集进行训练,理想的算法是在计算速度尽可能快的同时,使预测准确度尽可能高,泛用性尽可能强。因此,选择合适的机器学习算法对于获得更合理的预测结果至关重要。ECR催化剂筛选中常用的机器学习算法有神经网络、随机森林、高斯回归、支持向量机等。
训练完成的模型必须通过验证确认其具有良好的泛用性和准确性。在模型评估中,整个数据集通常分为训练集和测试集,训练集数据量占比一般为80%左右,测试集占比一般为20%[30]。需要注意的是,训练良好的机器学习模型也可能包含一些误差。其原因为在训练过程中存在数据噪声、离群数据、缺失数据等负面因素[12]。因此,模型只需要满足误差指标在一定限度内即可。评价机器学习模型训练好坏的指标有平均绝对误差(mean absolute error, MAE)、均方根误差(root mean square error, RMSE)等。实际过程中也会结合图像评判模型训练的好坏,常用真实值与预测值的对比图来直观展现机器学习训练效果。PEDERSEN等[31]利用高斯回归模型预测了高熵合金CO和H的吸附能,图7表示该模型的预测精度,绝大多数数据均位于±0.1 eV误差线内,表明模型取得了较好预测效果。
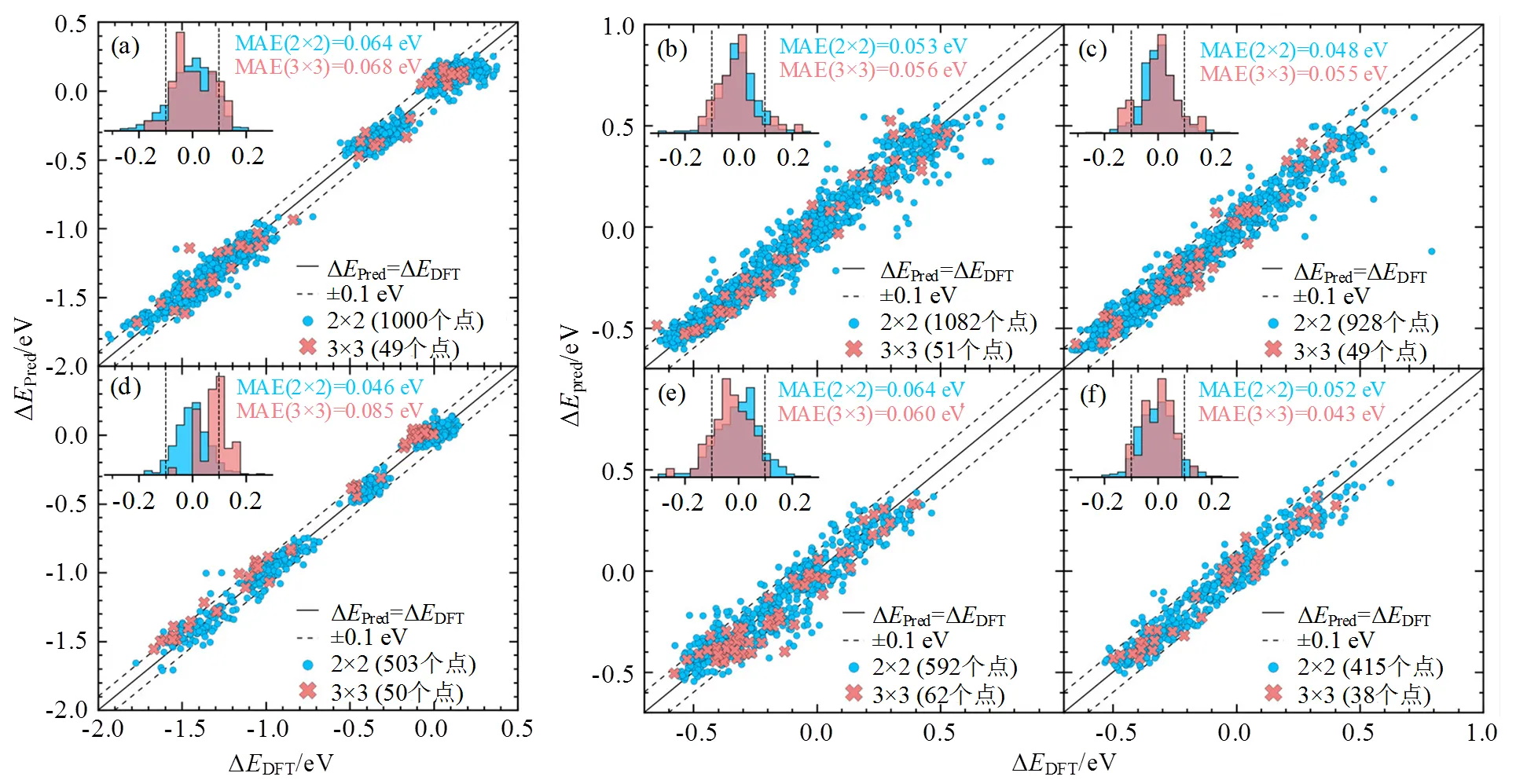
图7 DFT计算值与ML预测值对比结果[31]:(a)CoCuGaNiZn催化剂金属顶位点的CO吸附能;(b)CoCuGaNiZn催化剂的面心立方空位点的H吸附能;(c)CoCuGaNiZn催化剂六方最密堆积空位点的H吸附能;(d)AgAuCuPdPt催化剂金属顶位点的CO吸附能;(e)AgAuCuPdPt催化剂的面心立方空位点的H吸附能;(f)AgAuCuPdPt催化剂六方最密堆积空位点的H吸附能
4.3 机器学习筛选ECR催化剂
与传统量子化学计算方法相比,机器学习的优势是在预测催化性能和设计新型催化剂方面具有更快的速度和更低的计算成本[32]。在ECR催化剂设计过程中,机器学习可以对吸附能、d带中心、配位数等进行预测,这些值与CO2的转化和产物的形成密切相关。可对反应活性和选择性等进行预评估,从而加速新型高性能催化剂的设计与开发进程。目前,机器学习在ECR催化剂开发的应用包括催化剂筛选、催化剂组分优化、活性位点识别等。TRAN等[33]使用遗传算法回归模型搜索金属间晶体和表面,以寻找CO2电催化还原和析氢反应的最佳活性,筛选了具有不同表面位置的多种双金属材料,得到了最利于ECR反应的CO吸附能信息,最终筛选出131个金属表面,包含54种不同金属组合。CHEN等[28]通过极限梯度提升、遗传算法、k近邻回归、随机森林回归、支持向量机、梯度增强回归等6种机器学习模型,选择预测性能最好的极限梯度提升模型对单原子催化剂的CO2还原反应性能进行预测,如图8(a ~ c)所示。结果表明,在该研究框架下最有前景的掺杂原子是Co、Ni、Fe、Ir、V、Os、Sc、Y、Zr和Ti。筛选获得高活性的催化剂后,通过密度泛函理论和从头算分子动力学模拟,进一步确定催化剂的电化学稳定性,如图8(d)所示。活性位点的识别包括对Au纳米颗粒[34]和双金属纳米颗粒的活性面判定[35]等。

图8 机器学习预测ECR催化剂[28]:(a)特征的皮尔逊相关性系数;(b)特征重要性;(c)DFT与机器学习算法XGBR对ΔGCO的计算与预测结果;(d)设计材料的结构稳定性
5 结 论
对ECR催化剂设计与开发的理论方法进行了综述,包括密度泛函理论、溶剂化模型、计算电化学和机器学习四个方面,获得的结论如下:
(1)传统的DFT主要以LDA或GGA作为交换相关泛函,计算效率最高,可用于结构优化和金属体系的电子结构(例如态密度、原子电荷等)和各种热力学性质(能量、状态方程等)的计算,对于包含强相关电子的系统,需要采用DFT + U或杂化泛函等提高计算精度。
(2)CO2电催化还原必须考虑溶液的溶剂效应。典型的溶剂化模型包括显式溶剂化模型、隐式溶剂化模型和混合隐式−显式溶剂化模型等。其中,显式溶剂化模型最接近真实的溶剂,计算精度高,但其计算量也最大;隐式溶剂化模型采用溶剂表示为以介电常数为特征的极化介质,优点是节约计算时间和成本。混合隐式−显式溶剂化模型平衡了计算精度和时间成本。
(3)CHE模型尽管使用起来相对简单,但其无法真实描述决速步。此外,CHE模型在计算相对能量时存在固有误差。CEP模型考虑了电极电位变化的影响,能够可靠描述CO2自由基阴离子在金属表面的吸附,在CO2电催化还原中更具应用价值。AIMD是一种加速采样方法,可通过添加正高斯势加速探索系统的自由能。
(4)机器学习的过程主要包括数据收集、特征工程、算法选择和模型验证四个步骤,其核心步骤在于特征工程,不同算法的训练效果也存在较大差异。在催化剂设计中,机器学习已应用于催化剂性能预测、组分优化、活性位点识别等。
6 展 望
尽管ECR催化剂理论设计方法已经取得了长足进展,理论设计方法在ECR催化剂开发中仍存在诸多局限性,理论方法未来可能的发展方向主要包括:
(1)电催化还原电势中的能量计算仍不全面。动力学建模将DFT参数映射到宏观测量的同时,也将宏观测量的不确定性映射到测量方法。然而,DFT方法的不确定性映射到多尺度方法还处于起步阶段,需要改进现有统计热力学方法,将DFT计算的性质更精确地映射到宏观性质。
(2)尽管ECR催化剂理论设计的DFT计算利用隐式和显式溶剂化模型处理溶剂化的影响,但获得更准确的溶剂化吉布斯自由能仍有一定难度,继续改进溶剂效应处理方法对于提高计算方法的可预测性至关重要。
(3)机器学习的经典任务是加速常规任务的进程。目前,机器学习主要针对ECR催化剂的选择性和活性预测,对产物机理研究等方面的应用较少。实际上,机器学习作为一个加速工具,未来可能的方向包括研究反应机理、加速筛选复杂体系催化剂、加速构建高性能ECR反应体系、与先进实验分析技术结合等。
[1] HUA Z X, YANG Y J, LIU J. Direct hydrogenation of carbon dioxide to value-added aromatics[J]. Coordination chemistry reviews, 2023, 478: 214982. DOI: 10.1016/ j.ccr.2022.214982.
[2] WHIPPLE D T, KENIS P J A. Prospects of CO2utilization via direct heterogeneous electrochemical reduction[J]. The journal of physical chemistry letters, 2010, 1(24): 3451-3458. DOI: 10.1021/jz1012627.
[3] 华亚妮, 冯少广, 党欣悦, 等. CO2电催化还原产合成气研究进展[J]. 化工进展, 2022, 41(3): 1224-1240. DOI: 10.16085/j.issn.1000-6613.2021-2009.
[4] KOHN W, SHAM L J. Self-consistent equations including exchange and correlation effects[J]. Physical review, 1965, 140(4A): A1133-A1138. DOI: 10.1103/ PhysRev.140.A1133.
[5] PERDEW J P, ZUNGER A. Self-interaction correction to density-functional approximations for many-electron systems[J]. Physical review B, 1981, 23(10): 5048-5079. DOI: 10.1103/PhysRevB.23.5048.
[6] ANISIMOV V I, ZAANEN J, ANDERSEN O K. Band theory and mott insulators: hubbardinstead of stoner[J]. Physical review B, 1991, 44(3): 943-954. DOI: 10.1103/PhysRevB.44.943.
[7] DUDAREV S L, BOTTON G A, SAVRASOV S Y, et al. Electron-energy-loss spectra and the structural stability of nickel oxide: an LSDA+U study[J]. Physical review B, 1998, 57(3): 1505-1509. DOI: 10.1103/ PhysRevB.57.1505.
[8] PICKETT W E, ERWIN S C, ETHRIDGE E C. Reformulation of the LDA+method for a local-orbital basis[J]. Physical review B, 1998, 58(3): 1201-1209. DOI: 10.1103/PhysRevB.58.1201.
[9] BECKE A D. Density-functional thermochemistry. III. The role of exact exchange[J]. The journal of chemical physics, 1993, 98(7): 5648-5652. DOI: 10.1063/1. 464913.
[10] 苑琦, 杨昊, 谢淼, 等. 二氧化碳电还原反应的理论研究[J]. 物理化学学报, 2021, 37(5): 2010040. DOI: 10.3866/PKU.WHXB202010040.
[11] ZHONG M, TRAN K, MIN Y M, et al. Accelerated discovery of CO2electrocatalysts using active machine learning[J]. Nature, 2020, 581(7807): 178-183. DOI: 10.1038/s41586-020-2242-8.
[12] BRUNK E, ROTHLISBERGER U. Mixed quantum mechanical/molecular mechanical molecular dynamics simulations of biological systems in ground and electronically excited states[J]. Chemical reviews, 2015, 115(12): 6217-6263. DOI: 10.1021/cr500628b.
[13] SENN H M, THIEL W. QM/MM methods for biomolecular systems[J]. Angewandte chemie international edition, 2009, 48(7): 1198-1229. DOI: 10.1002/anie. 200802019.
[14] FRANCKE R, SCHILLE B, ROEMELT M. Homogeneously catalyzed electroreduction of carbon dioxide—methods, mechanisms, and catalysts[J]. Chemical reviews, 2018, 118(9): 4631-4701. DOI: 10.1021/acs.chemrev.7b00459.
[15] KLAMT A, SCHÜÜRMANN G. COSMO: a new approach to dielectric screening in solvents with explicit expressions for the screening energy and its gradient[J]. Journal of the chemical society, perkin transactions 2, 1993(5): 799-805. DOI: 10.1039/P29930000799.
[16] MARENICH A V, CRAMER C J, TRUHLAR D G. Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions[J]. The journal of physical chemistry B, 2009, 113(18): 6378-6396. DOI: 10.1021/jp810292n.
[17] KRONAWITTER C X, LESSIO M, ZHAO P, et al. Observation of surface-bound negatively charged hydride and hydroxide on GaP(110) in H2O environments[J]. The journal of physical chemistry C, 2015, 119(31): 17762-17772. DOI: 10.1021/acs.jpcc.5b05361.
[18] LESSIO M, DIETERICH J M, CARTER E A. Hydride transfer at the GaP(110)/solution interface: mechanistic implications for CO2reduction catalyzed by pyridine[J]. The journal of physical chemistry C, 2017, 121(32): 17321-17331. DOI: 10.1021/acs.jpcc.7b05052.
[19] NØRSKOV J K, ROSSMEISL J, LOGADOTTIR A, et al. Origin of the overpotential for oxygen reduction at a fuel-cell cathode[J]. The journal of physical chemistry B, 2004, 108(46): 17886-17892. DOI: 10.1021/jp047349j.
[20] MONTOYA J H, SHI C, CHAN K, et al. Theoretical insights into a CO dimerization mechanism in CO2electroreduction[J]. The journal of physical chemistry letters, 2015, 6(11): 2032-2037. DOI: 10.1021/acs. jpclett.5b00722.
[21] MATHEW K, KOLLURU V S C, MULA S, et al. Implicit self-consistent electrolyte model in plane-wave density-functional theory[J]. The journal of chemical physics, 2019, 151(23): 234101. DOI: 10.1063/1. 5132354.
[22] GARZA A J, BELL A T, HEAD-GORDON M. Mechanism of CO2reduction at copper surfaces: pathways to C2products[J]. ACS catalysis, 2018, 8(2): 1490-1499. DOI: 10.1021/acscatal.7b03477.
[23] ZHAO X H, LIU Y Y. Unveiling the active structure of single nickel atom catalysis: critical roles of charge capacity and hydrogen bonding[J]. Journal of the American chemical society, 2020, 142(12): 5773-5777. DOI: 10.1021/jacs.9b13872.
[24] GU G H, CHOI C, LEE Y, et al. Progress in computational and machine-learning methods for heterogeneous small-molecule activation[J]. Advanced materials, 2020, 32(35): 1907865. DOI: 10.1002/adma. 201907865.
[25] ZAGORAC D, MÜLLER H, RUEHL S, et al. Recent developments in the Inorganic Crystal Structure Database: theoretical crystal structure data and related features[J]. Journal of applied crystallography, 2019, 52(5): 918-925. DOI: 10.1107/S160057671900997X.
[26] GRAŽULIS S, CHATEIGNER D, DOWNS R T, et al. Crystallography Open Database - an open-access collection of crystal structures[J]. Journal of applied crystallography, 2009, 42(4): 726-729. DOI: 10.1107/ S0021889809016690.
[27] JAIN A, ONG S P, HAUTIER G, et al. Commentary: the materials project: a materials genome approach to accelerating materials innovation[J]. APL materials, 2013, 1(1): 011002. DOI: 10.1063/1.4812323.
[28] CHEN A, ZHANG X, CHEN L T, et al. A machine learning model on simple features for CO2reduction electrocatalysts[J]. The journal of physical chemistry C, 2020, 124(41): 22471-22478. DOI: 10.1021/acs.jpcc. 0c05964.
[29] WEI J Z, YANG Y J, LIU J, et al. Design and screening of transition-metal doped chalcogenides as CO2-to-CO electrocatalysts[J]. Journal of CO2utilization, 2022, 64: 102165. DOI: 10.1016/j.jcou.2022.102165.
[30] NOBLE W S. What is a support vector machine?[J]. Nature biotechnology, 2006, 24(12): 1565-1567. DOI: 10.1038/nbt1206-1565.
[31] PEDERSEN J K, BATCHELOR T A A, BAGGER A, et al. High-entropy alloys as catalysts for the CO2and CO reduction reactions[J]. ACS catalysis, 2020, 10(3): 2169-2176. DOI: 10.1021/acscatal.9b04343.
[32] LAMOUREUX P S, WINTHER K T, TORRES J A G, et al. Machine learning for computational heterogeneous catalysis[J]. ChemCatChem, 2019, 11(16): 3581-3601. DOI: 10.1002/cctc.201900595.
[33] TRAN K, ULISSI Z W. Active learning across intermetallics to guide discovery of electrocatalysts for CO2reduction and H2evolution[J]. Nature catalysis, 2018, 1(9): 696-703. DOI: 10.1038/s41929-018-0142-1.
[34] CHEN Y L, HUANG Y F, CHENG T, et al. Identifying active sites for CO2reduction on dealloyed gold surfaces by combining machine learning with multiscale simulations[J]. Journal of the American chemical society, 2019, 141(29): 11651-11657. DOI: 10.1021/jacs. 9b04956.
[35] ULISSI Z W, TANG M T, XIAO J P, et al. Machine-learning methods enable exhaustive searches for active bimetallic facets and reveal active site motifs for CO2reduction[J]. ACS catalysis, 2017, 7(10): 6600-6608. DOI: 10.1021/acscatal.7b01648.
Recent Progress of Theoretical Design Methods for CO2Electrocatalytic Reduction Catalysts
WANG Qinghua1, XIAO Yiyang2, YANG Yingju2,†, LIU Jing2, BAI Hongcun3
(1. Hefei Power Generation Co. Ltd., CHN Energy Investment Group, Hefei 230026, China; 2. State Key Laboratory of Coal Combustion, School of Energy and Power Engineering, Huazhong University of Science and Technology, Wuhan 430074, China; 3. State Key Laboratory of High-efficiency Utilization of Coal and Green Chemical Engineering, School of Chemistry and Chemical Engineering, Ningxia University, Yinchuan 750021, China)
CO2electrocatalytic reduction (ECR) is a promising technology for CO2utilization. The key problem depends on the development of highly efficient catalysts. The theoretical method can effectively guide and accelerate the design of efficient ECR catalyst. This review introduces the theoretical methods of ECR catalyst design from four aspects: density functional theory (DFT), solvent effect, electrochemical computational model and machine learning. DFT, DFT + U and hybrid functional can effectively calculate the energy and electronic properties of ECR reaction system and predict the performance of catalyst. The selection of explicit solvation model, implicit solvation model and mixed model should be considered to investigate the solvent effect in ECR reaction. In the electrochemical calculation of ECR, the constant electrode potential (CEP) model is more effective than the computational hydrogen electrode (CHE) model in describing the energy change of CO2reduction process. Machine learning enables the performance prediction, active site design and component optimization of ECR catalysts with high efficiency and low cost. Finally, an outlook on the rational design methods of catalysts for CO2electrocatalytic reduction is provided.
CO2electrocatalytic reduction; density functional theory; electrochemical calculation; machine learning
2095-560X(2023)06-0524-10
TK16
A
10.3969/j.issn.2095-560X.2023.06.006
2023-08-22
2023-09-28
中央高校基本科研基金项目(2019kfyRCPY021);湖北省创新群体项目(2023AFA039);省部共建煤炭高效利用与绿色化工国家重点实验室开放课题项目(2022-K46)
杨应举,E-mail:yangyingju@hust.edu.cn
王清华, 肖一杨, 杨应举, 等. 二氧化碳电催化剂理论设计方法研究进展[J]. 新能源进展, 2023, 11(6): 524-533.
: WANG Qinghua, XIAO Yiyang, YANG Yingju, et al. Recent progress of theoretical design methods for CO2electrocatalytic reduction catalysts[J]. Advances in new and renewable energy, 2023, 11(6): 524-533.
王清华(1974-),男,硕士,工程师,主要从事火电厂节能与减碳研究。
杨应举(1990-),男,博士,讲师,主要从事二氧化碳热/电催化转化利用、氢能绿色制取、碳基能源转化利用、燃烧污染物控制等研究。
