基于生物信息学的急性髓系白血病患者预后风险模型的构建与验证
2024-01-04张斯娜马书杰
张斯娜,马书杰
1.淄博市疾病预防控制中心性病艾滋病防制所(山东淄博 255020)
2.河南理工大学医学院(河南焦作 454003)
急性髓系白血病(acute myeloid leukemia,AML)是一种未成熟髓系造血干细胞异常克隆性恶性肿瘤,具有高度异质性,其主要特征是克隆性造血干细胞或祖细胞(hematopoietic stem and progenitor cells, HSPCs)分化和凋亡障碍。AML可导致克隆性HSPCs 在骨髓中恶性增殖和聚集,从而影响骨髓正常造血功能[1-2]。AML 的高度异质性与多种因素有关,如基因组突变、表观遗传学异常和基因表达异常等[3]。全球范围内,AML发病人数从1990 年的63 840 例增加到2017 年的119 570 例,发病率在过去的28 年间增加了87.3%[4]。化疗、生物制剂和干细胞移植是AML的主要治疗方式[5-7]。尽管AML 的治疗取得了重大进展,但化疗药物毒性可能导致急性和危及生命的并发症。AML 通常预后不良,患者中位生存时间仅为8.5 个月,2 年和5 年总生存率(overall survival, OS)均低于35%,仅35%~40%的年轻患者(<60 岁)和5%~15%的老年患者(≥60 岁)在化疗后可存活5 年以上[8-9]。随着分子生物学的迅速发展,基因组学、蛋白质组学和生物信息学分析方法已被用于开发新的个性化治疗策略,研究相关生物分子的功能,收集临床数据基因组匹配的新趋势信息是改善AML 患者预后的有效方法[10-11]。尽管许多研究分析了AML 的基因组变异,但基因组变异与AML 分子机制间的关联仍不明确。因此,需要进一步探索更有效的AML早期诊断和预后方法。AML 的基因表达谱已被证明在诊断不同的细胞遗传学亚型、发现新的AML亚类和预测预后方面具有重要价值[12-13]。基因组分析是筛选潜在的疾病诊断和预后生物标志物的一种有效方法,本研究基于公共数据库的基因表达谱数据集构建并验证AML 患者预后风险模型,识别AML 患者的预后生物标志物。
1 资料与方法
1.1 数据来源与差异表达基因筛选
基于癌症基因组图谱(The Cancer Genome Atla, TCGA)数据库(https://gdc.xenahubs.net/download/TCGA-LAML/Xena_Matrices/TCGALAML.htseq_fpkm.tsv.gz),获取AML 基因数据(151 例)作为病例组。基于基因型-组织表达(Genotype-Tissue Expression, GTEx) 数据库(https://toil.xenahubs.net/download/TCGatargettex_rsem_gene_fpkm.gz),获取正常骨髓样本(70 例)作为对照组。利用Wilcoxon 秩和检验分析AML样本基因数据,筛选|logFC|>2 且错误发现率(false discovery rate, FDR)<0.05 的差异表达基因(differentially expressed genes, DEGs)。采用单因素Cox 回归分析,筛选DEGs 中与AML 生存相关基因,以P<0.01 为差异有统计学意义。
1.2 风险评分模型构建与特征基因验证
151 例AML 样本中,剔除临床特征信息缺失和随访时间为0 的样本后,最终纳入132 例,将132 例AML 样本按照1 ∶ 1 的比例随机分成两组,分别作为训练集和测试集。在训练集中,通过单变量Cox 比例风险回归分析mRNA 的表达与患者OS 之间的关系。在构建风险预测模型前,采用随机生存森林算法对AML 预后相关的差异基因进行筛选。在随机生存森林监督分类算法中,采用迭代过程缩小基因集。随机森林的树数(ntree)为1 000,将每一步输入节点数的平方根设为单个分类树的每个节点随机选取的预后相关的差异mRNA 的大小,对袋外数据样本进行泛化误差估计。为了选择极少最重要的基因变量,可依据变量的重要性(per-mutation variable important,VIMP)对变量进行筛选,VIMP 值越大表明其预测能力越强。风险预测评分模型是基于AML 预后相关的差异基因而构建,通过其估计回归系数加权如下:
其中,N 为预后相关的差异基因数量,Expi为预后相关的差异基因的表达值,Coei为单变量Cox 回归分析中预后相关差异基因的估计回归系数。模型构建后,模型中涉及的特征基因通过以下方式进一步验证:①对模型高低风险组的生存分析,最佳的截断表达值由“survminer”R 包的“surv_cutpoint”函数确定。使用Kaplan-Meier和log-rank 方法在训练集中进行生存分析,再使用AML 数据和临床信息在测试集中验证该模型。②在训练集和测试集中使用“survivalROC”R 软件包进行时间依赖的受试者工作特征(receiver operating characteristic, ROC)曲线分析,并计算1 年、2 年和3 年AML 患者生存率的ROC 曲线下面积(area under the curve, AUC)。通过决策曲线分析量化不同时间概率下的净收益,从而确定风险评分的临床实用性,决策曲线分析通过“rmda”R 软件包进行。基于所有样本的基因表达,使用“Rtsne”R 软件包进行t-SNE 分析,探索不同组的分布。
1.3 分层分析
在全集中,对AML 患者总生存率显著相关的临床特征数据进行分层分析,其中年龄以60岁为截断值,≥60 岁为老年组,<60 岁为年轻组。
1.4 功能富集分析
为了阐明与风险评分相关的生物学功能和途径,高风险组和低风险组之间的DEGs 被用于进行基因本体(Gene Ontology, GO)富集分析。利用“clusterProfiler”R 包进行分析,以校正后P值小于0.05 为差异有统计学意义。
2 结果
2.1 AML患者临床特征
训练集、测试集和全集中AML 患者的临床特征见表1。
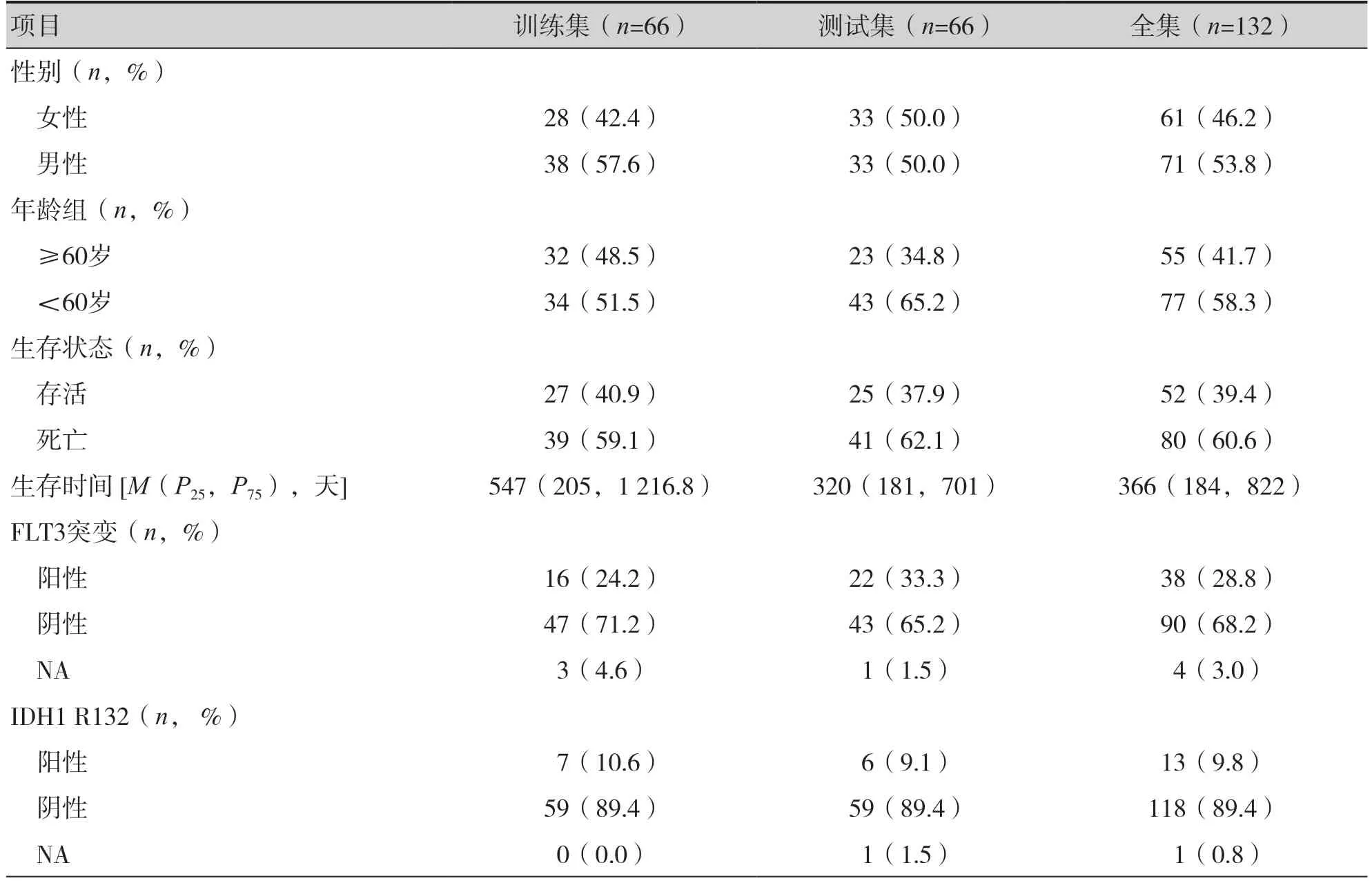
表1 AML患者的临床特征Table 1.Clinical characteristics of patients with AML
2.2 差异表达基因鉴别
基于AML 样本和正常骨髓样本数据,共获取19 022 个基因的mRNA 测序数据,通过基因差异显著性分析筛选出2 270 个DEGs(图1)。通过单变量Cox 回归分析,得到335 个AML 预后相关特征基因(P<0.01)。
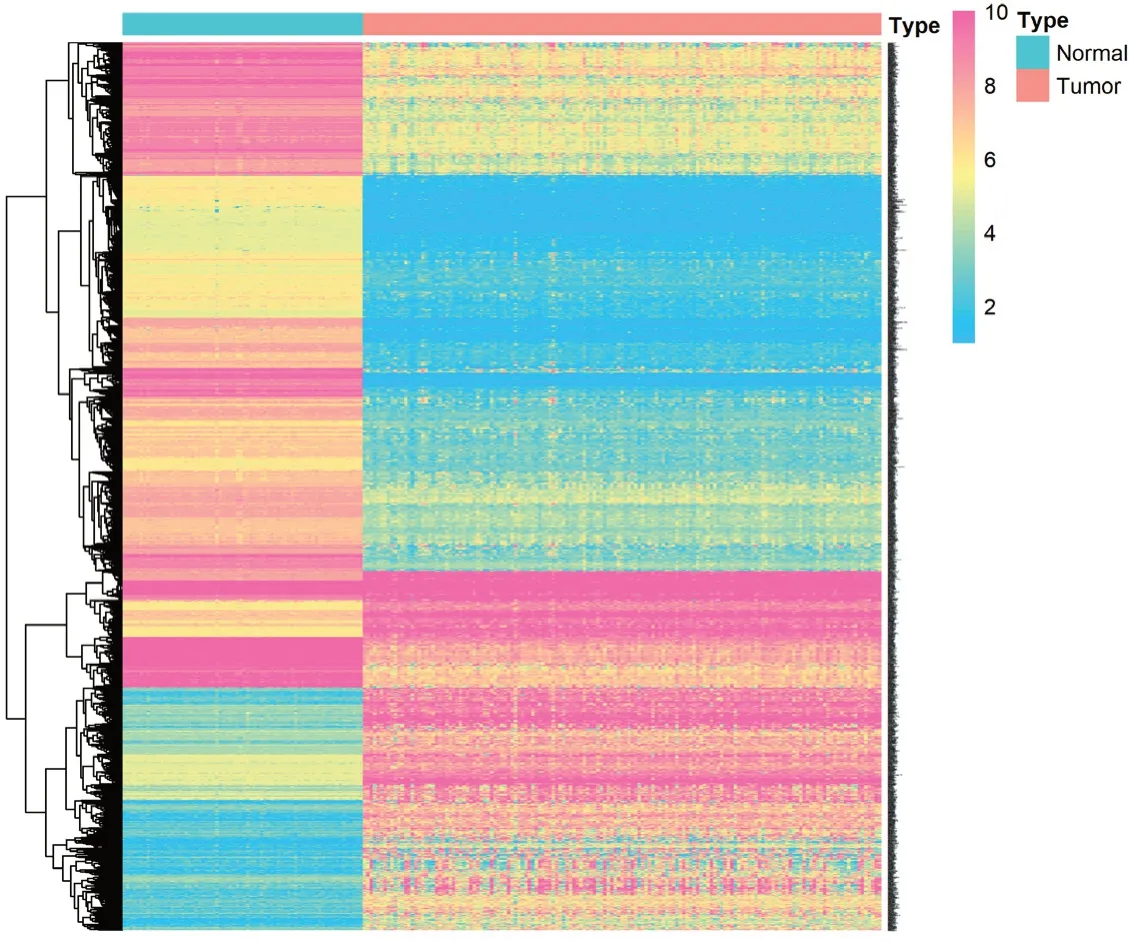
图1 差异基因表达热图Figure 1.Heatmap of differential genes expression
2.3 特征基因筛选
为了进一步缩小预后基因的数量,通过随机森林算法对335 个与AML 患者OS 显著相关的DEGs 进行分析,在每一步中,根据通过置换测试使用袋外样品对每个基因的重要分数进行估计,丢弃不重要的基因,并根据每一步中的排列重要评分,得到与AML 患者OS 显著相关的5 个基因标志物 (ACOT7、SFXN3、SLC29A2、HINT2和ZMAT1),见图2-A。
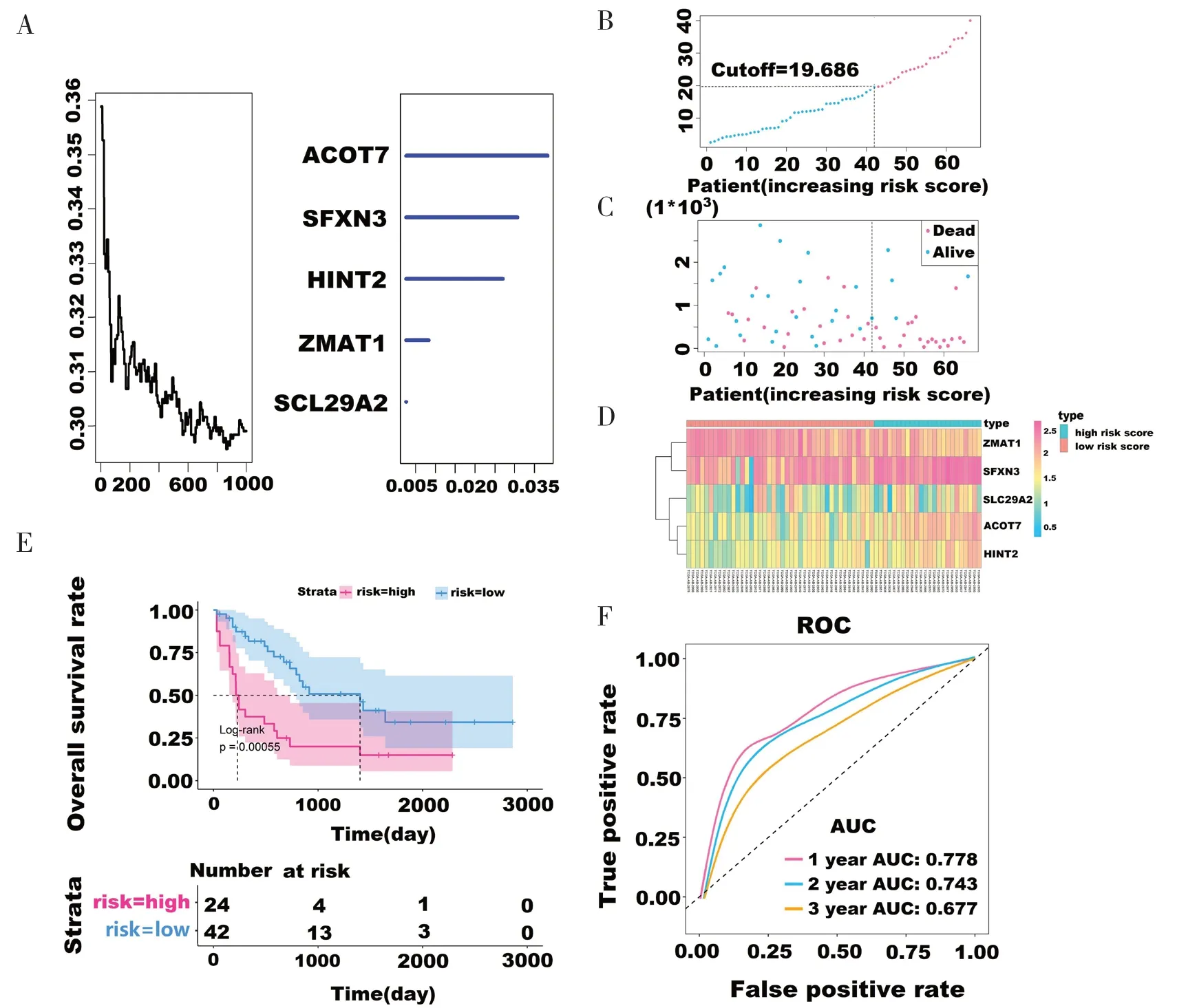
图2 特征基因风险评分分析Figure 2.Risk scoring analysis of characteristic genes
风险评分模型构建如下:风险评分=(-0.959 24×ZMAT1表达值)+(0.525 21×SFXN3表达值)+(1.060 22×HINT2表达值)+(0.669 68×SLC29A2表达值)+(0.079 19×ACOT7表达值)。基于风险评分模型计算训练集每个AML 患者的风险评分,根据评分将患者分为高风险组(n=24)和低风险组(n=42),使用cutoff=19.686 作为风险临界值。图2-B、图2-C、图2-D 展示了66 例AML 患者的预后评分、生存状态和肿瘤mRNA 表达的分布,根据5 个特征基因的预后评分值进行排序。在5 个基因中,4 个基因(ACOT7、SFXN3、SLC29A2和HINT2)与预后高风险相关 [风险比(hazard ratio, HR)>1],1 个基因(ZMAT1)为保护性因素(HR <1)。预后评分高的基因倾向于表达高风险的mRNA,而预后评分低的基因倾向于表达保护性mRNA,见图2-C 和图2-D。Kaplan-Meier 生存分析和对数秩检验表明,高风险组和低风险组的总体生存率有显著差异,高风险组患者总生存期明显短于低风险组(中位生存期3.79 年 vs.0.57 年,P<0.001),见图2-E。5 个基因生存预测的时间依赖ROC 曲线分析显示,1 年、2 年、3 年的AUC 值分别为0.778、0.743、0.677,曲线接近左上角,具有良好的敏感性和特异性,表明该预测模型具有较高的准确率,见图2-F。
2.4 特征基因验证
为了测试5 个特征基因在预测AML 患者总生存期中的预后价值,在测试集中测试5 个特征基因。通过使用相同的风险评分模型,测试集的66 例患者被分为高风险组(n=33)和低风险组(n=33)。结果显示,高风险组患者的总生存期明显短于低风险组患者(中位生存期0.71 年vs.2.5 年,P<0.001),见图3-A。时间依赖的ROC 曲线分析对5 个特征基因的生存预测的AUC值在1 年时达到0.775,2 年时达到0.746,3 年时达到0.696,见图3-B。进一步将5 个特征基因应用于整个数据集的患者,以验证其预测价值,来自训练集的相同风险评分模型和中位风险截止标准将全集的132 例AML 患者分为高风险组(n=66)和低风险组(n=66)。高风险组患者的总生存期明显短于低风险组患者(中位生存期0.68 年 vs.2.58 年,P<0.001),见图3-C。在132 例AML患者全集中验证5 个特征基因的ROC 曲线在1 年、2 年、3 年的AUC 值分别为0.836、0.777、0.694,见图3-D。
2.5 特征基因的预后价值与临床变量的独立性
如表2 所示,多变量Cox 回归分析结果表明,经年龄、性别和各分子分析异常测试结果调整后,5 个特征基因仍与AML 患者总生存率存在显著相关性。当控制其他临床变量时,在训练集中,高风险组相对于低风险组总生存率的风险比为3.47[95%CI(1.80,6.70),P< 0.001],在测试集和全集中,高风险组相对于低风险组总生存率的风险比分别为3.25 [95%CI(1.71, 6.19),P<0.001]和3.39 [95%CI(2.12,5.43),P<0.001]。年龄与三组数据集AML 患者的总生存率显著相关,因此根据年龄进行分层分析,所有患者被分为年轻组(n=77)和老年组(n=55),再根据风险评分模型,将年轻组和老年组患者分为高风险组和低风险组。如图4-A 和图4-B 所示,生存分析表明,在年轻组和老年组中,高风险组的总体生存时间明显短于低风险组(P<0.001)。
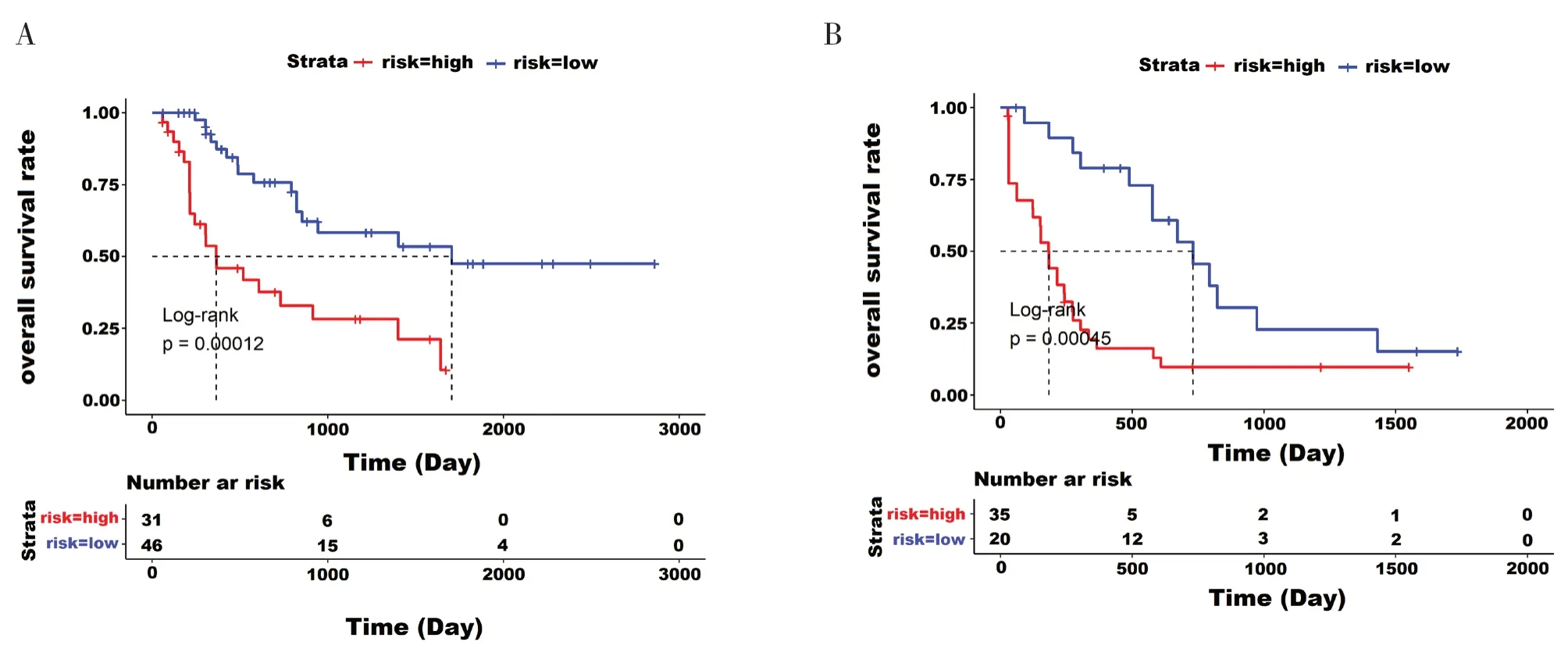
图4 两组总体生存率的估计Figure 4.Estimation of overall survival rates for two groups
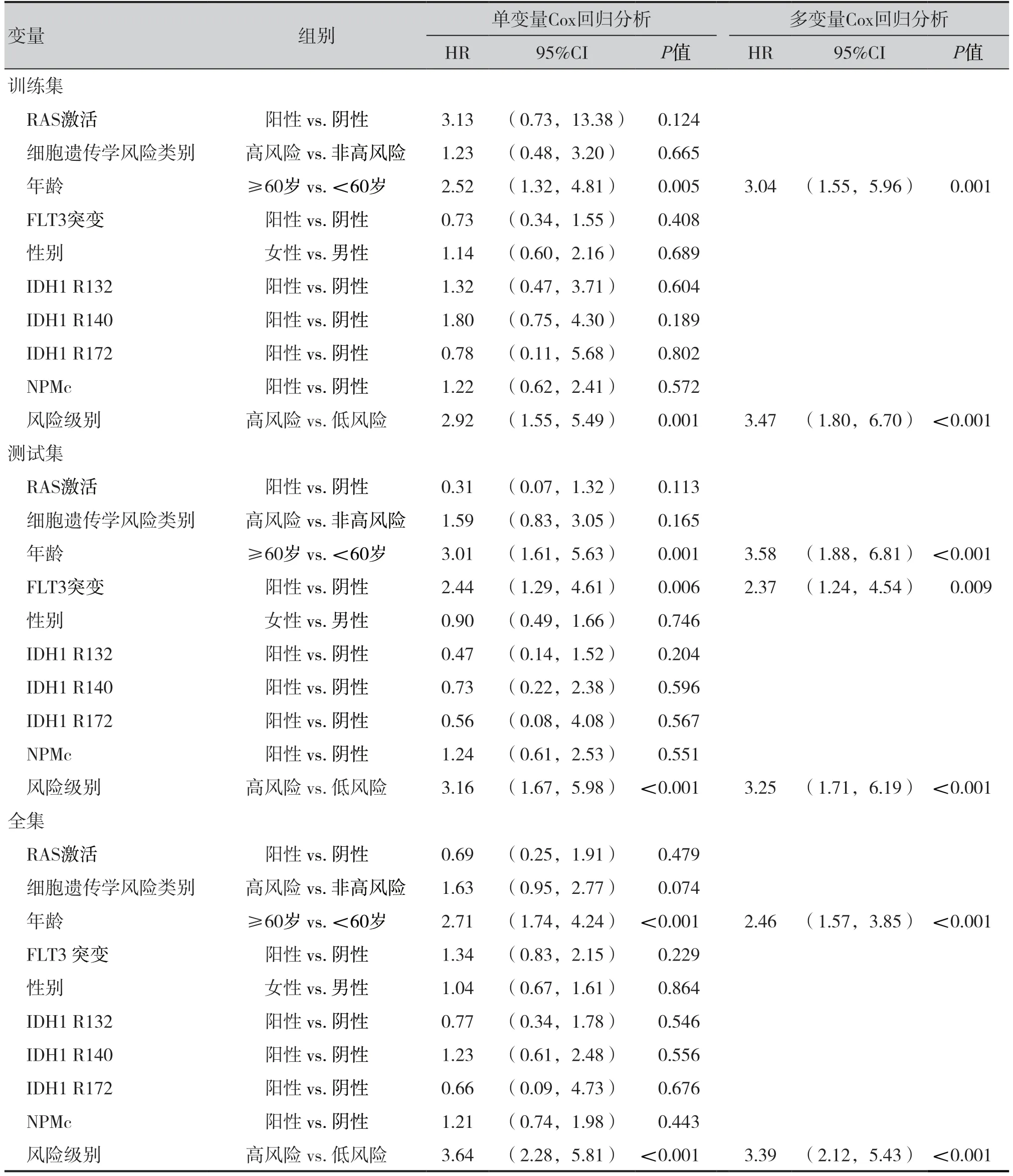
表2 单变量和多变量Cox回归分析结果Table 2.Results of univariate and multivariate Cox regression analysis
2.6 特征基因的性能评估
t-SNE 分析表明,不同风险组的患者分布在两个方向(图5-A)。5 个特征基因组合风险评分的决策曲线分析表明,风险评分预测AML 患者1 年、2 年和3 年的生存情况表现出比极端曲线更高的净获益,而1 年的阈值范围更大,认为1 年风险评分表现更优,见图5-B。

图5 特征基因的性能评估Figure 5.Performance evaluation of characteristic genes
2.7 功能富集分析
如图6-A 所示,在整个数据集中,DEGs 富集几种与代谢过程相关的途径,如固醇代谢过程、胆固醇代谢过程、类固醇代谢过程、仲醇代谢过程、脂质代谢过程和酒精代谢过程(校正后P<0.05);在许多免疫相关的生物过程中也明显富集,如白细胞迁移、T 细胞分化和巨噬细胞迁移(校正后P<0.05);DEGs 在凋亡相关的生物过程中也明显富集,如凋亡信号通路的负调控、线粒体凋亡途径和线粒体释放细胞色素C途径(校正后P<0.05)。图6-B 展示了各途径所富集到的基因。
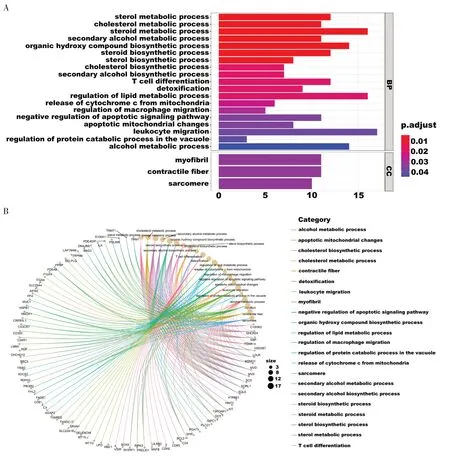
图6 GO富集分析Figure 6.GO enrichment analysis
3 讨论
在最近的研究中,许多AML 相关的癌基因和肿瘤抑制基因已被确定。例如,Zhou 等发现HSPG2过表达与AML 患者的不良预后相关,AML 患者中HSPG2的表达水平显著上调,完全缓解后则下调,而在复发时再次升高[14]。Li 等研究表明,SLC38A1是AML 的重要预后和预测指标,SLC38A1高表达患者的NPM1基因突变率较低,不良风险核型发生率较高,SLC38A1表达水平高的患者总生存期显著缩短[15]。Wróbel 等发现IL-17F多态性与AML 的诱导密切相关[16]。尽管发现了一些与AML 预后和诊断相关的基因,但其在AML 中的作用仍有待探究。AML 的预后部分由遗传因素驱动,多种基因的结合有助于提高预后预测的准确性。本研究中,AML 患者的临床数据和基因表达数据来自TCGA 数据库,以鉴定与预后相关的基因。通过单变量Cox 分析和随机生存森林分析选择生存相关特征基因,最终通过多变量Cox 回归分析构建了基于5 个预后相关特征基因的风险评分模型。
在本研究筛选出的5 个特征基因中,仅ZMAT1被认为是低风险且与AML预后相关基因。目前尚未发现ZMAT1在AML 中的研究报告,但在胰腺导管腺癌中有过报道,研究发现ZMAT1的下调与胰腺导管腺癌不良的临床病理特征和低生存率相关,ZMAT1通过诱导SIRT3/p53信号通路在胰腺导管腺癌中起到抑癌因子的作用,其中ZMAT1促进SIRT3基因的转录,随后上调p53的表达,并参与调节胰腺导管腺癌的细胞周期进程和凋亡[17]。Zhang 等研究发现高水平的ACOT7对AML 患者的预后有显著影响,ACOT7高表达组的无事件生存期(event-free survival, EFS)和OS显著降低[18]。ACOT7的表达水平是影响AML 患者预后的独立因素,ACOT7的高表达水平在男性患者中更常见,且表达水平与FAB 亚型以及细胞遗传学之间可能存在潜在相关性[18]。SLC29A2也是ENT2,有研究表明肝细胞癌(hepatocellular carcinoma, HCC)组织中SLC29A2的上调与晚期、血管浸润与患者生存率不良显著相关[19]。有研究推测SLC29A2的异常上调可能改变细胞核苷酸合成和核苷酸池平衡,从而导致癌症基因组不稳定,进而为肿瘤提供生长优势[20]。由于SLC29A2是一种已知的腺苷转运体,而从缺氧组织中释放的腺苷刺激血管内皮生长因子的释放,再结合内皮细胞上的受体,刺激内皮细胞增殖、迁移和肿瘤血管生成[20]。SFXN3和HINT2已被证明分别在口腔鳞状细胞癌和肝细胞癌中作为癌基因[21-22]。Murase 等认为口腔鳞状细胞癌患者血清抗SFXN3自身抗体水平显著升高,血清抗SFXN3-autoAb水平与原发肿瘤大小轻度相关,但在早期癌症中水平稍高,SFXN3蛋白在癌巢之间的基质成纤维细胞中表达,也在鳞状上皮的基底层中表达,治疗后血清抗SFXN3自身抗体水平的变化与临床肿瘤负荷相关[21]。Zhou 等研究表明HINT2在HCC组织中的表达明显低于邻近的非肿瘤组织,且HINT2低表达HCC 患者复发的可能性更高,可作为HCC 复发的一个预后指标[22]。鉴于在其他类型癌症中的重要作用,这些特征基因可用于预测AML 患者的生存率。基于5 个特征基因的风险评分模型,低风险和高风险患者的存活率有显著差异,高风险患者比低风险患者存活时间明显更短。
基于不同风险组间的DEGs 进行了GO 富集分析,发现许多免疫相关的生物过程和途径得到了富集,说明AML 可能与肿瘤免疫有密切联系的推测具有一定合理性。基于5 个特征基因的风险评分模型初步用于AML 患者的生存预测,并进一步证明了这些特征基因在AML 生存中的重要作用,可作为更好地评估AML 患者生存和预后的新方法。此外,风险评分模型在测试集及整个数据集中得到进一步验证,表明该模型在AML患者生存预测中具有良好的可重复性。由于流行病学的限制,本研究未能分析风险评分与AML患者生存率之间的具体关系,未来有待进一步深入探究。
综上所述,本研究确定了5 个与AML 预后密切相关的特征基因,并构建和验证了基于5 个特征基因的风险评分模型,该模型可用于预测AML 患者的生存期,为AML 患者的个性化风险评估、治疗和预后提供参考。
