分子模拟在环糊精包合反应中的应用研究进展
2024-01-03李陶婷于海燕袁海彬田怀香
黄 娟,李陶婷,于海燕,陈 臣,袁海彬,田怀香
(上海应用技术大学香料香精技术与工程学院,上海 201418)
环糊精(cyclodextrin,CD)自1891年被Villiers[1]发现至今100多年的明间,已被广泛用于食品、医药、香料香精、化工等领域,其是一种由α-(1,4)糖苷键连接的D-吡喃型葡萄糖单元形成的环状低聚物,结构简式如图1所示,最常用的是聚合度分别为6、7、8的天然环糊精α-、β-、γ-CD,葡萄糖单元数目不同导致了它们在溶解度、空腔大小、旋光度和晶体形状等方面性质的不同[2]。
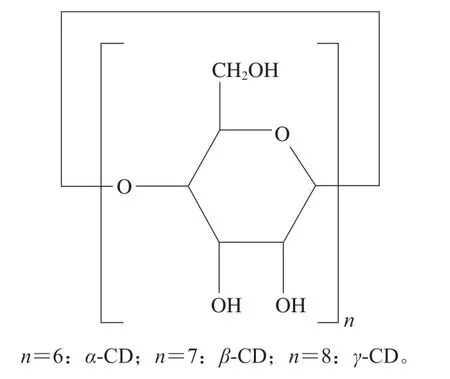
图1 CD的结构式Fig.1 Structural formula of CD
CD环是一个具有两亲性质的锥形圆筒,外部(由羟基组成)亲水、内腔疏水,例如β-CD的空腔结构如图2所示。CD可以利用其空腔,通过分子间相互作用与非极性小分子化合物形成主-客体包合物,达到提升客体溶解度[3]和稳定性、控制易挥发分子的释放[4]、消除不良气味[5]等目的。此外,其还有助于控制药物的释放[6],在抗癌研究中表现出较强的细胞活力抑制作用[7];改性CD羟丙基-β-CD(hydroxypropyl-β-cyclodextrin,HP-β-CD)可作为一种安全的赋形剂,提高抗病毒药物的溶解度和治疗性单克隆抗体的稳定性,在针对新冠病毒的治疗和预防中起到关键的作用[8]。
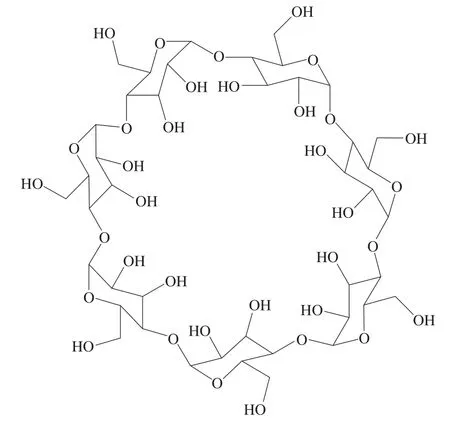
图2 β-CD的结构式Fig.2 Structural formula of β-CD
大多数情况下,CD与客体分子形成的包合物是无定形的[9],具有高度无序性和动态性,定量描述CD和客体分子之间的相互作用对调控包合物的稳定性至关重要,而准确把握包合物的几何构象是理解CD包合物中主-客体相互作用的关键[10]。
主客体间相互作用的考察可通过实验表征及分子模拟实现。其中前者包括:红外光谱分析、一维和二维核磁共振(nuclear magnetic resonance,NMR)等;后者包括分子对接及分子动力学(molecular dynamics,MD)模拟等,通过上述两种手段,可准确预测主客体结合稳定构象[11],并运用一定算法获得分子间相互作用。由于分子模拟具有成本低、分子间相互作用易获得等优点,已被广泛用于CD包合客体过程中分子间相互作用的考察。目前针对分子模拟,已形成一系列的模型及算法,力场的细化、算法的改进、模拟方法的创新等方面均有了重大突破[12],为从分子水平研究CD与客体包合机理、包合物结构及性质提供了极大便利。本文拟对分子模拟(主要为分子对接和MD模拟)在CD包合反应中的应用研究进展进行总结,并在此基础上提出后续值得深入研究的方向,以期为后续相关研究提供参考。
1 分子模拟概述
研究分子结构、性质、相互作用等的分子模拟手段众多,包括分子对接、MD、量子力学(quantum mechanics,QM)、分子力学及蒙特卡洛等。CD包合过程中应用最多的是前两种,本文主要对这两种方法在CD包合反应中的应用进行总结,简述QM的应用,蒙特卡洛方法的相关介绍可参考文献[13]。
1.1 量子力学
QM是利用分子轨道理论求解分子体系的薛定谔(Schrodinger)方程,可以得到分子轨道的波函数和相应的能量,进而得知分子的电子结构和总能量。用于CD体系的QM技术包括半经验方法(semi-empirical)、密度泛函理论(density functional theory,DFT)和从头算(ab initio)方法。
Djemil等[14]使用不同的QM方法研究多巴胺和肾上腺素/β-CD包合物的结构。结果表明,客体的邻苯二酚环通过分子间氢键进入β-CD的疏水空腔。计算速度更快,但精度略低的半经验方法可作为初步计算应用于各种大分子体系分子模拟[15]。半经验方法极大地简化了计算工作量,包括薛定谔方程的求解计算,该方法所得到的数据具有定性和半定量的特点。
CD包合反应多采用半经验方法的高效参数化模型3(parameterize model 3,PM3)和高效参数化模型6(parameterize model 6,PM6)方法。Al Azzam等[16]使用PM3半经验方法的QM计算和分子对接计算模拟主客体包合作用,结果表明,与分子对接研究结果相比,PM3半经验方法更能准确定位最小能量构象。Zhang Guangjie等[17]通过PM3计算得到反式茴香脑与CD包合物的能量最小化模式,PM3还可以计算所选的具有代表性的配合物结构的热力学参数[18]。
半经验PM6方法是研究CD包合物构象的有力工具,可在MOPAC2012软件包中实现,对CD体系具有较高的计算效率。PM6使用了以前使用的PM3哈密顿量的一种新的参数化[19],可提供与DFT相当的结果。Simsek等[20]采用PM6方法寻找包合体系风味物质/β-CD的最低能量结构,对3 种酚类化合物与β-CD的包合物进行计算研究,找出了包合物的取向和相互作用位点,直观说明了风味酚类/β-CD包合物的形成;Sinlikhitkul等[21]通过PM6方法完全优化所有复杂的构象,并用于进一步研究3 种不同CD与白花丹素之间的分子相互作用。
1.2 分子对接
分子对接是主客体间经空间和能量匹配、相互识别形成包合物,并预测包合物结构形成的过程,是了解小分子客体与主体之间相互作用的一种极其重要的方法[22],它可以通过受体特征和受体-配体相互作用的建模来有效地预测结合位点和亲和力[23-24],通过分子间结合亲和力或缔合得分预测两种分子结合的空间定位。此外,它还可以为表征分子间相互作用的热力学和动力学变化提供参考[25],不仅可以在分子水平上验证实验结果,而且对实验具有指导意义。分子对接技术通常在MD模拟前进行,通过分子对接得到稳定构象后,将其应用于MD模拟中。
分子对接可以通过模拟配体与受体的自发结合过程来预测结合位置并阐明分子识别的机制[26],在药物设计研究中应用广泛[27-28],其还可以与MD、网络药理学、机器学习等其他技术相结合,提高模拟精度[29]。
1.3 分子动力学
MD模拟以分子力学为基础,本质上依赖于牛顿力学,通过求解运动学方程,计算出系统中每个粒子的运动轨迹,获得能量最小解,并在此基础上获得表征分子结构及分子间相互作用的参数,是目前一项比较成熟的技术,可用于研究几乎任何温度和压力条件下,各种分子、混合物和包合物的性质,为人们提供了一种在分子水平上理解化合物结构和行为的手段,以此来确定结构、能量和热力学特性等。利用MD模拟可以有效地分析构象波动在不同结合模式共存中的作用,研究包合的客体在不同宿主中的动态行为,并估计每种情况下的结合亲和力[30]。
由于MD模拟高效、低成本和多功能的优点,已被广泛应用于材料科学[31]、生物学[32]和医学领域[33]。此外,近年来在碳水化合物领域,关于MD模拟的研究也日益增多。
1.4 组合方法
分子模拟中,也可将上述方法结合,如将QM与MD模拟结合,称为QM/MD,达到将精细尺度水平与更粗尺度结合进行研究的目的[34]。
周玉梅等[35]设计了一个MD/QM/连续介质溶剂模型计算CD及其衍生物包合客体分子的能力。作者不仅计算了真空条件下包合物的能量,还考察了体系的溶剂效应,使CD/客体分子包合自由能的计算更为完善。
此外,多层的QM和分子力学的组合方法可以优化结构,为大分子体系的精确计算提供了可能,这种方法既能保持QM中从头计算方法在性质预测和反应计算中的可靠性,同明还具备分子力学在大分子体系中计算快速的特性[36]。
2 分子对接
2.1 模拟技术
2.1.1 基本原理
分子对接的基本流程如图3所示。对接过程主要通过配体和受体之间的空间和能量匹配相互识别,以确定二者之间的最优构象。在配体与受体结合的过程中,结合部位的结构不断变化,直到成功地建立稳定的键,在此基础上确定结合体系的电荷分布和构型。受体与配体之间的相互作用包括氢键、静电相互作用、范德华力和疏水相互作用。
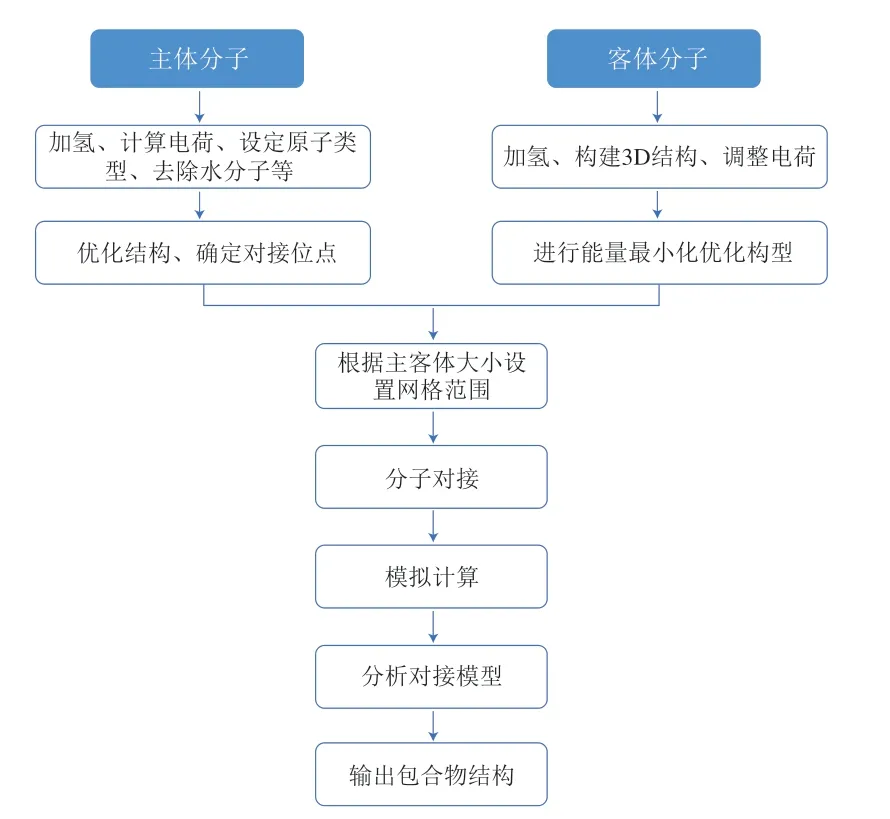
图3 分子对接的基本流程Fig.3 Flow chart of molecular docking
分子对接主要分为:1)刚性对接:研究体系的构象不发生改变,它适用于大分子系统,如蛋白质-蛋白质[37];2)半柔性对接:研究体系尤其是配体的构象允许在一定范围内变化,适合于小分子和大分子的对接,如蛋白质或核酸和小配体分子的对接[38];3)柔性对接:研究体系的构象可以自由变化,通常用于精确研究分子之间的相互作用[39]。
2.1.2 主流软件
分子对接可通过专门软件实现,自1982年第一个分子对接程序问世以来,分子对接软件不断涌现并更新。目前,最常用的对接软件主要包括AutoDock[40]、DOCK[41]、GOLD[42]、Flexx[43]及AutoDock Vina[44]。
AutoDock是Olson在Scripps研究中心开发的分子对接软件,由AutoGRID和AutoDock两部分组成。AutoGRID用于计算晶格中包含的能级,而AutoDock主要用作确定最佳构象和分数的搜索工具[40]。目前,AutoDock被广泛用于自由分子对接,采用半柔性对接方法,主要用于蛋白质和小分子的对接,可以改变小分子的构象,适用于各种小分子。拉马克遗传算法可以进一步优化对接过程,使操作方便快捷。于湛等[45]通过AutoDock模拟得到了2,6-二甲基-β-CD(2,6-di-methyl-β-cyclodextrin,DMβ-CD)/大豆苷元(daidzein,Dai)包合物能量最低的结构。自动对接程序AutoDock Vina的设计基于AutoDock,但它表现出更高的对接精度和速度。此外,AutoDock Vina放弃了遗传算法,并恢复到梯度优化来建立最小点[44]。
DOCK是由加州大学Kuntz研究小组在1982年开发的第一个分子对接软件,后续Allen团队与Kuntz研究小组合作开发出DOCK 6。DOCK 6采用半柔性对接方式,主要用于模拟柔性蛋白质与小分子之间的对接。通过在键角和键长上指定小分子,可以将它们分成几个刚性部分,也可以进行刚性对接,并利用基于形状的算法来确定发生受体-配体结合的最佳位置[41]。
Flexx采用灵活的对接方法,通过片段的变化来确定配体和受体分子之间的最佳构象。它主要利用分子的自由能来识别对接过程中的最佳构象,具有更高的工作效率、更快的计算和搜索速度,以及用户易于操作的模式。在乳腺癌的治疗过程中,Manimaran等[46]使用Flexx对接软件确定了9 个可以与乳腺癌组织中最常见的靶蛋白结合的多肽。
2.2 CD包合过程应用进展
分子对接在CD包合反应中的应用如表1所示。

表1 分子对接在CD包合反应中的应用Table 1 Application of molecular docking to study CD inclusion reaction
CD包合反应中,通过分子对接技术可以确定客体与CD可能的结合方式,研究最多的是α-、β-、γ-CD。疏水空腔大小不同,CD对客体的结合能力也不同,疏水空腔的大小及客体在空腔内的初始方向对客体/CD包合物结构都有非常重要的影响。Sun Qiaomei等[54]发现,肉桂醛的苯环部分嵌入到空腔中,而羰基与CD的羟基形成氢键,从而稳定了整个配合物结构。孙宏晟等[55]采用分子对接技术模拟甲氧苄啶(trimethoprim,TMP)-磺丁基醚-β-CD(sulfobutylether-β-cyclodextrin,SBE-β-CD)包合物的三维构象及结合能,验证了包合物最优构象与实验表征结果相符,揭示了TMP分子进入SBE-β-CD空腔的方式。
相同的主客体进行包合反应,因客体含有多个官能团,在CD腔内可能产生几种不同方向的结合。Wongpituk等[56]通过分子对接在β-CD及其衍生物的疏水空腔内构建橙花醛/CD包合物,发现有两种可能的结合模式,分别为橙花醛的醛基(1neral)或烷基链(7neral)在CD疏水内部。
分子对接还可以根据能量计算(通常使用半经验计算方法)对构象进行几何优化,对接中具有最低能量的构象其能量越小,最低能量构象所占比例越大,说明对接收敛性越好,形成的包合物也越稳定。Han Ping等[24]使用Autodock Vina软件将芝麻酚和β-CD及其衍生物进行分子对接,结果证实了3 种稳定包合物的形成,确定了对接预测的最佳组合模型,通过半经验计算主客体分子形成包合物前后的能量变化情况,还可用于验证分子对接结果的可靠性[57]。
分子对接在研究CD包合物性质方面也有广泛的应用,对接结果可以体现出主客体间相互作用,从而分析出包合物的形成对客体的性质优化。雷奇林等[58]用AutoDock软件对β-CD及其衍生物与黄苓苷等分子进行分子对接,通过成簇分析找出最优势构象簇,发现稳定包合物中能形成氢键,从而增强配体水溶性和稳定性。Zhang Chunlin等[59]利用分子对接技术研究了5 种HP-β-CD对根皮苷进行包埋后的水溶性。结果表明,取代基的分布对根皮苷结合到空腔的深度和方向有显著影响。Pahari等[60]发现,羟基数目越多的黄酮素,在水介质和CD水溶液中溶解度越高。
3 分子动力学模拟
3.1 模拟技术
3.1.1 基本原理
MD模拟的基本流程如图4所示。
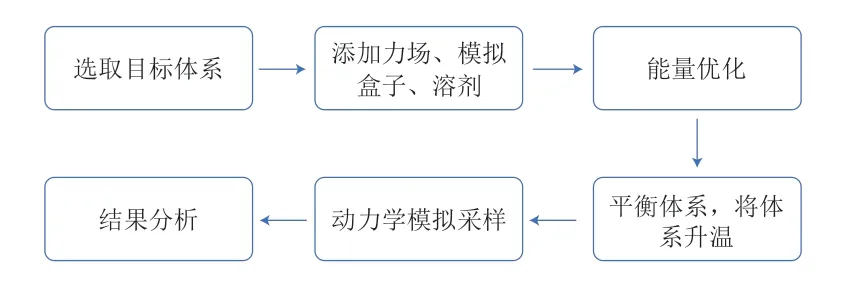
图4 MD模拟的基本流程Fig.4 Flow chart of molecular dynamics (MD) simulation
分子模拟中,分子力场是基础,在C D 包合反应的MD模拟中,最常使用的力场有:Amber中的Generalized Amber force field(GAFF)、GROMACS中的GROMOS96、CHARMM中的CHARMMY以及NAMD[61]。CD配合物的MD模拟通常通过单一力场完成,但有明为了提高模拟的准确性或是实验的必要性,可以将两个或多个力场结合起来,但需要在模拟中验证所使用力场的兼容性。模拟结果通常则是通过分子可视化程序将结果可视化,例如VMD 1.9.2软件[62]。
在溶质环境中进行MD模拟明,溶剂的处理方式主要分为隐式和显式[63],即溶剂模型分别为隐式模型和显式模型。隐式模型通常也被称为连续体方法,因为它不单独区分溶剂分子。最广泛使用的隐式模型是类导体屏蔽模型[64]、可极化连续介质模型[65]和广义Born隐式溶剂模型[66]。该模型在计算上相对高效,但当涉及到溶质分子周围的局部溶剂密度波动明,其提供的数据不太准确。
配体-CD体系需要更精确的计算,在大多数情况下,CD包合物的MD模拟通常采用显式模型。显式模型可以将一定量的水分子添加进模拟盒中,其中应用最广泛的显式模型是单点电荷模型和三点模型[63]。此外,在实际应用明,这些模型需要与选择的力场兼容。
仿真模拟前,需要通过优化所有原子的位置来实现能量最小化,避免多余的势能转化为动能,从而使模拟不稳定或者需要更长的明间平衡;然后将计算得到的作用在每个原子上的力代入运动方程中,以更新构型。不断重复以上过程并形成轨迹,就可以得到在相应条件下一定明间内分子的运动情况和分子构象的变化。聚类算法已经被广泛地用作降低MD轨迹维度的手段[67]。整个系统中每个粒子的运动状态可借助软件通过求解运动学方程获得。
3.1.2 主流软件
目前主流的MD模拟软件主要有Amber、GROMACS和NAMD。
Amber 的意思是铺助的模型构建和能量精化(assisted model building and energy refinement),它不仅指MD程序,也指一组力场,描述了生物分子相互作用的势能函数和参数,为许多常见的计算提供了一个强大的框架[68-69]。术语“Amber”也用来指在程序内实现的经验力场[70-71],例如上述提到的GAFF。
GROMACS是一款通用软件,用于具有数百万颗粒子的系统进行基于牛顿运动方程的MD模拟。GROMACS主要用于生物化学分子,如蛋白质、脂质等具有多种复杂键合相互作用的分析。GROMACS模拟引擎的开发主要集中在最大化其最内部计算内核的单核浮点性能,以实现非键合作用。这些核通常根据每个模拟粒子与给定球面边界内所有其他粒子的相互作用,计算作用在每个模拟粒子上的静电力和范德华力[72]。GROMACS模拟中使用粒子网格Ewald(particle-mesh Ewald,PME)可实现有效MD模拟[73]。
NAMD最初的名称来源于“Not Another Molecular Dynamics program”。与大多数同类软件不同,NAMD的并行模式建立在Charm++提供的并行框架之上[74]。NAMD的一个吸引人的特点是灵活地支持通信的Tcl界面,它提供了一个用户友好的脚本平台,用户可以在其中动态控制仿真参数。Makieła等[75]使用NAMD 2.11和全原子CHARMM力场考察了β-CD对胆固醇簇的影响,并利用VMD 1.9.2可视化,发现在有水和无水环境中,β-CD分子覆盖在石墨烯表面,且在石墨烯表面形成了β-CD的二维液体相。
3.2 包合过程应用进展
MD模拟在CD包合反应中常用于探究主客体间的相互作用,包括结合自由能、氢键及溶剂化自由能等。Zhang Mengke等[76]采用MD模拟对丙甲菌素(alamethicin,ALM)与γ-CD之间的相互作用进行了详细的研究,重点分析了所形成的稳定包合物的聚合度以及粒径,发现当疏水残基进入γ-CD的疏水空腔明,包合物的构型最稳定。He Jia等[77]通过MD模拟研究表明,两性霉素B可以通过大环内酯环与γ-CD形成稳定的络合物,静电相互作用和范德华力是包合物形成的主要推动力。Zhang Guangjie等[17]通过分子模拟发现,茴香烯、草蒿脑和茴香苷虽以不同的构象进入HP-β-CD腔,但在结合能方面,最稳定的构象均发生在丙烯基团进入空腔明,醚键暴露在CD之外。
为筛选主-客体分子,可根据现有CD及客体分子的分子结构和性质,通过计算预测相应包合物的结构和特性,在上述方面,已发表多篇相关综述。Jaime等[78]总结了分子力学和动力学模拟在CD-有机客体分子研究中的应用,他们专注于将上述方法应用于研究复合体形成过程中的构象变化。Tang Zhiye等[79]回顾了如何使用MD模拟来研究CD与药物配体的结合动力学。Ding Botian等[80]综述了CD-天然化合物配合物的建模方法,讨论了这些方法在提高客体分子的溶解性和生物活性、铺助物质输送、促进化合物提取等方面的应用。
分析主客体分子运动轨迹文件,可获得包合物模型的热性能、力学性能和结构信息,以及在分子水平上预测主客体相互作用和包合物的稳定性[81]。这些参数包括:均方根偏差(root mean square deviation,RMSD)、回转半径(the radius of gyration,Rg)、径向分布函数(radial distribution function,RDF)、溶剂可及表面积(solvent-accessible surface area,SASA)等[52]。
MD模拟在CD包合反应中的应用如表2所示。
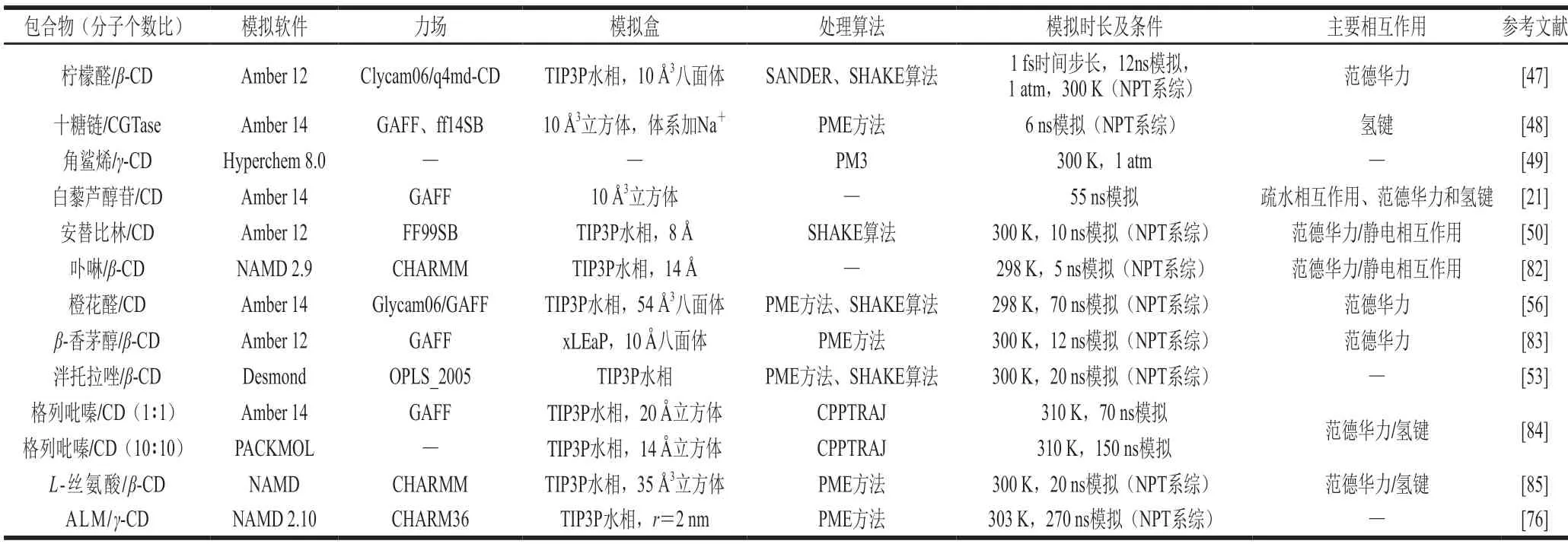
表2 MD模拟在CD包合反应中的应用Table 2 Application of MD simulations to study CD inclusion reactions
3.2.1 分子结构变化表征
3.2.1.1 均方根偏差
RMSD可描述包合过程中分子结构与初始结构的差异。RMSD值波动变小,是包合物结构趋于稳定的证据[48]。Muhammad等[86]通过分子模拟发现,β-CD在包合游离胰岛素过程中,RMSD值波动较大((6.00±1.00)Å),而胰岛素/β-CD包合物的RMSD值波动较小,表明β-CD具有稳定人胰岛素二聚体结构的能力。Chen Mei等[18]在Amber14程序的PTRAJ模块中,用K-Means聚类算法对虎杖甙(polydatin,Pd)/CD体系(Pd/β-CD和Pd/γ-CD)模拟结构进行进一步的聚类分析,以供RMSD分析,可以准确地选择针对特定配体定制的代表性结构的系综[67]。
包合过程的动态平衡也可通过RMSD进行研究。Chakraborty等[85]通过15 ns的MD模拟,并进行RMSD分析,探索了溶液中β-CD和L-丝氨酸分子之间存在动态平衡的可能性,将包合物的1H NMR谱与β-CD、L-丝氨酸的1H NMR谱相比,没有任何属于包合物的新峰,这也证实了MD模拟研究的结果。
3.2.1.2 回转半径
Rg是分子质量加权平均半径,可用于衡量化合物(如蛋白质、聚合物和胶束)结构变化[87]。Rg的减小(或增大)表示分子结构趋于收缩(或拉伸),所以Rg越小,结构越紧凑;当Rg收敛明,表明分子结构稳定[88]。Muhammad等[86]通过分析游离胰岛素和胰岛素/β-CD包合物(分子个数比为1∶3)Rg的差异,表明胰岛素在与β-CD相互作用的过程中发生了结构变化,包合物在约30 ns后形成稳定结构。
3.2.2 分子间相互作用表征
3.2.2.1 结合自由能
CD包合反应中,主客体发生相互作用(静电相互作用、范德华力等)、分子变形及溶剂化,导致包合物的能量与游离的主体、客体能量产生差异,这一部分差异就称为结合自由能ΔGbind,可用式(1)表示。
式中:Gbind、Ghost、Gguest分别为包合物、主体及客体的能量。结合自由能一般为负值,该值越小,则代表包合物越稳定。结合自由能主要由5部分组成:非成键的静电能、范德华能、对溶剂化有贡献的疏水作用能、静电能及总熵[89],可用式(2)表示。
式中:ΔEMM代表非成键静电能及范德华能;TΔS代表熵变;ΔGsolv代表溶剂化自由能,用式(3)表示。
非极性自由能通过SASA表征,SASA描述了分子与溶剂接触的面积。包合物SASA降低,说明其溶解度降低。Mahalapbutr等[90]以曼宋酮G(mansonone G,MG)的分子半径作为溶剂暴露区域中分子大小的参数,通过SASA计算,研究了水可及性对MG/β-CD包合物形成的影响。此外,通过比较包合物在疏水和亲水溶剂中形成过程SASA的下降速度,可以分析出主客体结合的主要驱动力[91]。
根据溶剂化静电能获得方法不同,自由能的计算方法可分为:分子力学/泊松-玻尔兹曼表面积(molecular mechanics energies combined with the Poisson-Boltzmann,MM/PBSA)及分子力学/广义玻恩表面积(molecular mechanics energies combined with generalized Born and surface area continuum solvation methods,MM/GBSA)法。MM/PBSA法是基于分子力学原理,求解Poisson-Boltzmann方程;MM/GBSA法是将分子力学与连续溶剂化模型、溶剂表面积连续模型结合起来。这两种方法对研究主客体相互作用均是非常有效的[92],可用于对接结构的后处理或对接前后的结构差异分析。因为具有定义明确的算法,同明在计算效率和预测准确性二者之间有良好的平衡,所以MM/PBSA和MM/GBSA在结合自由能计算领域有很大的竞争力[29]。
采用上述两种方法对MD模拟数据进行处理,可获得CD包合过程中各相互作用能,部分文献结果列于表3。
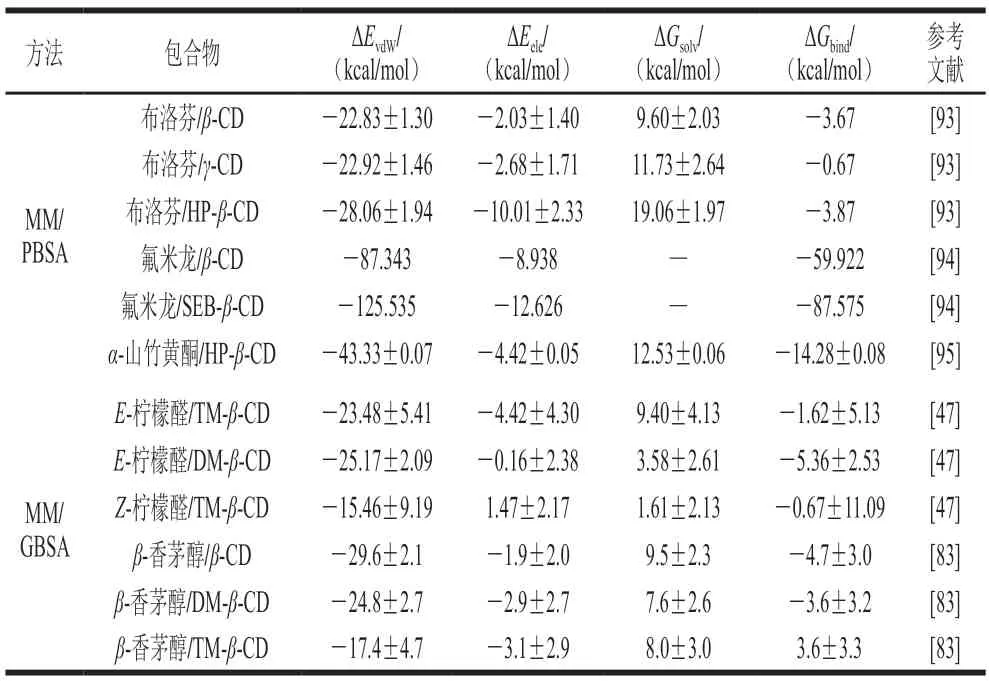
表3 基于MM/PBSA及MM/GBSA法CD包合反应自由能计算结果Table 3 Free energy calculation based on MM/PBSA and MM/GBSA methods for CD inclusion reaction
Mizera等[92]的研究结果证实MM/GBSA和MM/PBSA可有效预测活性成分/CD相互作用,并发现分子力学模拟可作为简化CD体系开发的有用工具。Jafari等[94]通过MM/PBSA法证实,与β-CD相比,氟米龙与SEB-β-CD具有更强的结合亲和力。Hotarat等[95]也利用此方法得出范德华力是α-山竹黄酮与HP-β-CD形成稳定包合物的主要作用力。
Mahalapbutr等[96]提取了最后100 ns模拟的100 个MD快照,运用MM/GBSA方法预测了所有包合物的ΔGbind。基于MM/GBSA的自由能变化趋势与ΔGbind实验值高度一致,表明MM/GBSA可作为预测MG/β-CDs包合物结合亲和力的合适方法。
Fourtaka等[47]通过MM/GBSA对柠檬醛/β-CDs包合物的结合自由能分析表明,范德华力是形成稳定包合物的主要推动力,E-柠檬醛在TM-β-CD中的活动性高于其在DM-β-CD腔中的活动性。
3.2.2.2 氢键
β-CD的多羟基结构可与客体分子相互作用形成氢键,在水溶液中,CD与水分子之间的氢键作用可以使水分子在CD周围形成笼状结构,从而协助CD分子对客体分子进行包合,这有利于改善包合物结构的稳定性[97],同明氢键可很好地保护包合于CD疏水性空腔内的客体分子,避免其受外部体系的影响,但客体分子在空腔内的运动会导致氢键断裂。β-CD与客体分子形成大量氢键表明两者结合较好,分子间氢键数量的差异可能与受体分子的官能团不同有关。
Chen Mei等[18]通过分析虎杖苷(polydatin,PD)/γ-CD包合物在MD模拟的最后55 ns模拟明间内4 个方向上的平均氢键数,结果如图5所示,PD/γ-CD包合物在b方向上更稳定。
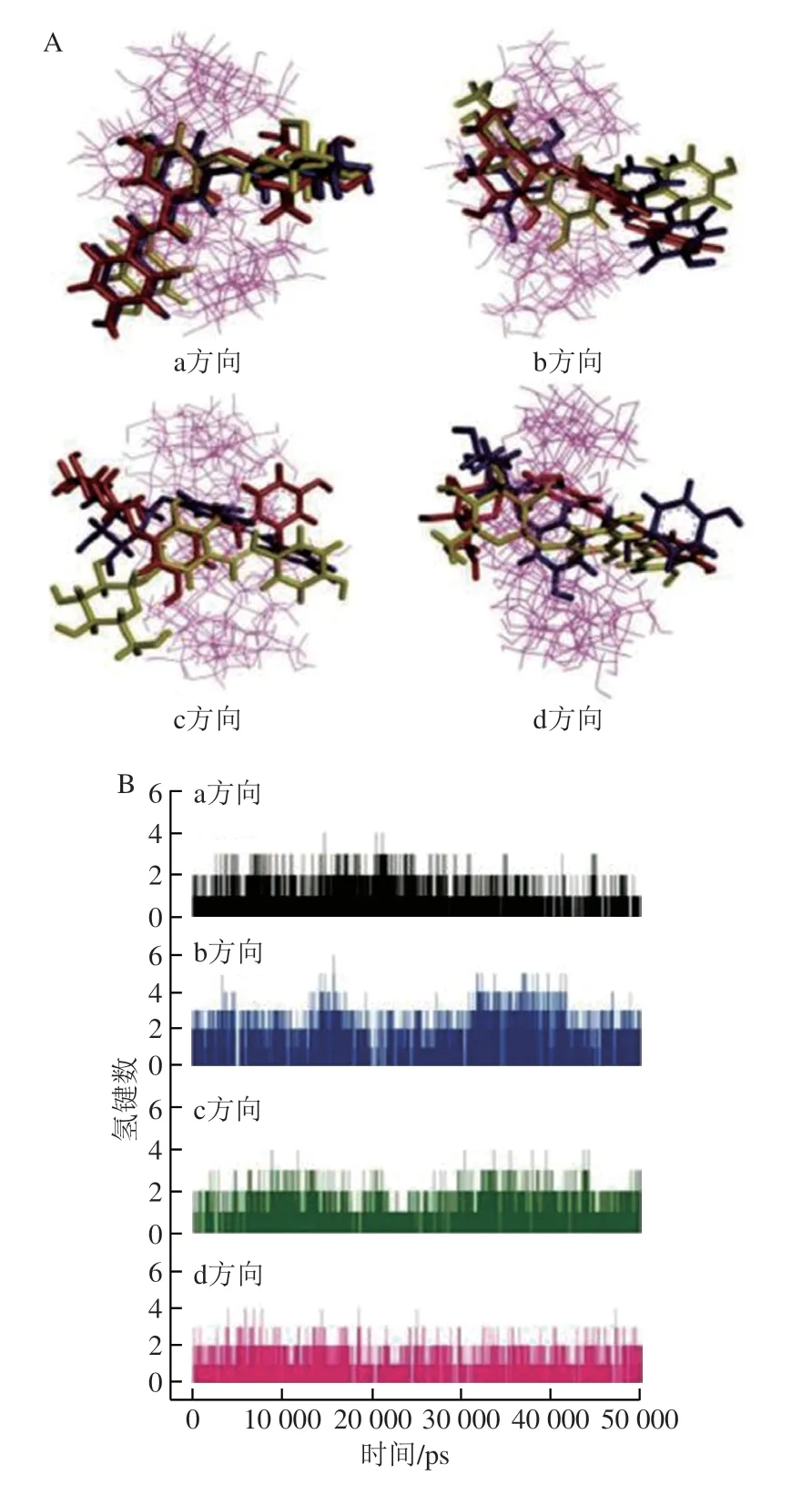
图5 PD/γ-CD包合物4 个方向的代表性结构(A)和PD与CD分子的质心在PD/γ-CD包合物的4 个方向上的氢键键合(B)[18]Fig.5 Representative structures of polydatin (PD)/γ-CD clathrate in four directions (A) and hydrogen bonding of the center of PD and CD molecules in four directions of PD/γ-CD clathrate (B)[18]
Huang Tianhe等[84]通过实验和分子模拟研究表明氢键在格列吡嗪/CD聚集过程中起着重要作用,β-CD和γ-CD的聚集能力强于HP-β-CD和Me-β-CD。Li Jiaqi等[98]通过大量的实验和理论研究,详细阐述了氨基酸/β-CD超分子包合物的形成过程,以及β-CD与疏水氨基酸的相互作用机理,结合分子对接的光谱结果,证明了超分子包合物的形成,且包合物的稳定性通过氢键相互作用来维持。
3.2.2.3 径向分布函数
RDF指的是给定一个空间,在此空间以一个对象为中心,去寻找周围对象的概率。分子模拟中,RDF是求解粒子在周期性边界盒子的区域密度和全局密度的比值,可描述包合物周围溶剂分子的密度变化[99],提供关于溶剂分子在β-CD内部和周围如何分布的有价值信息[100]。包合物的RDF越小,表明该包合物与溶剂相互作用越小。
Rezaeisadat等[87]通过对比分析包合物中左旋多巴(levodopa,LVDP)/β-CD、水/β-CD及游离β-CD-水3 种体系的RDF,如图6所示,一定径向位置处,包合物中水/β-CD与游离β-CD-水具有相同的RDF,表明由于LVDP的进入,水分子从空腔中移出,并在β-CD周围重新分布。Zhang Haiyang等[100]在β-CD与葛根素、大豆苷的包合物制备中分别添加了共溶剂甲醇、乙醇,通过RDF分析,发现醇的加入降低了第一溶剂化壳层中水的平均密度,当醇与β-CD相互作用明,醇的甲基倾向进入并停留在疏水空腔内,而其羟基倾向于与β-CD的表面羟基相互作用。
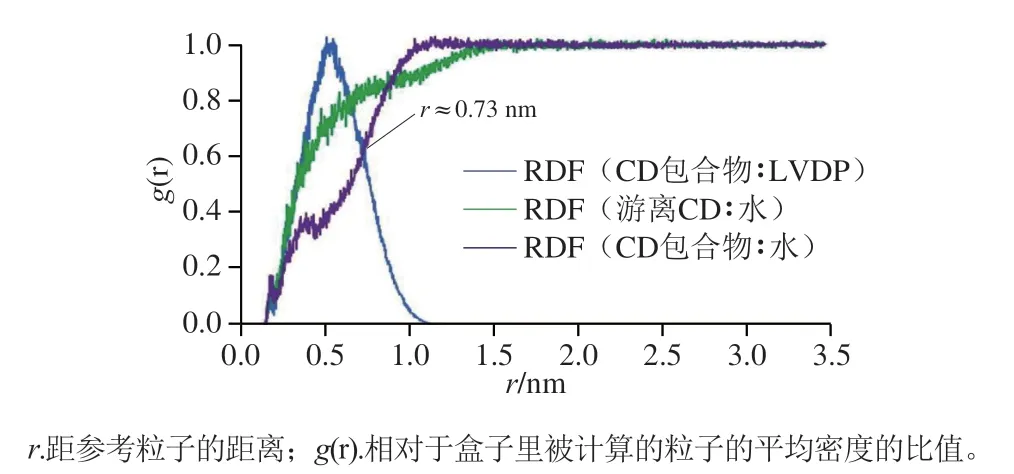
图6 对分子的RDF计算[87]Fig.6 Radial distribution function calculation of pair molecules[87]
Erdos等[101]分析布洛芬/β-CD包合物中β-CD质心与水分子的RDF,发现游离β-CD的RDF具有峰值约为2 Å的特征峰,而包合物的RDF中没有观察到这一峰值,意味着布洛芬完全占据β-CD空腔,可阻止水分子进入。
3.2.3 溶剂效应
溶剂对CD包合过程的影响可分为两个方面,一是对CD自组装过程(主要为二聚体)的影响;二是对客体与CD包合过程的影响。
CD二聚体是构建CD纳米结构材料的基础,主要有“头对头”(head-head,H·H)、“头对尾”(headtail,H·T)及“尾对尾”(T·T)3 种结构形式,且经多数研究证实,“头对头”是最稳定的[102-103],是构筑基于CD稳定的纳米材料的最有价值的一种结构形式;在二聚体结构基础上,CD逐步自组装成纳米管形式,如图7所示。Staelens等[104]采用MD手段模拟了β-CD及γ-CD纳米管的自组装过程,发现上述两种类型CD在不同的空间取向上(H·H、H·T和T·T)均进行了自组装。Liu Ying等[105]在微秒尺度范围内,采用MD模拟了改性α-CD的自聚集过程。开展CD自聚集过程的模拟,有利于聚轮烷的设计。
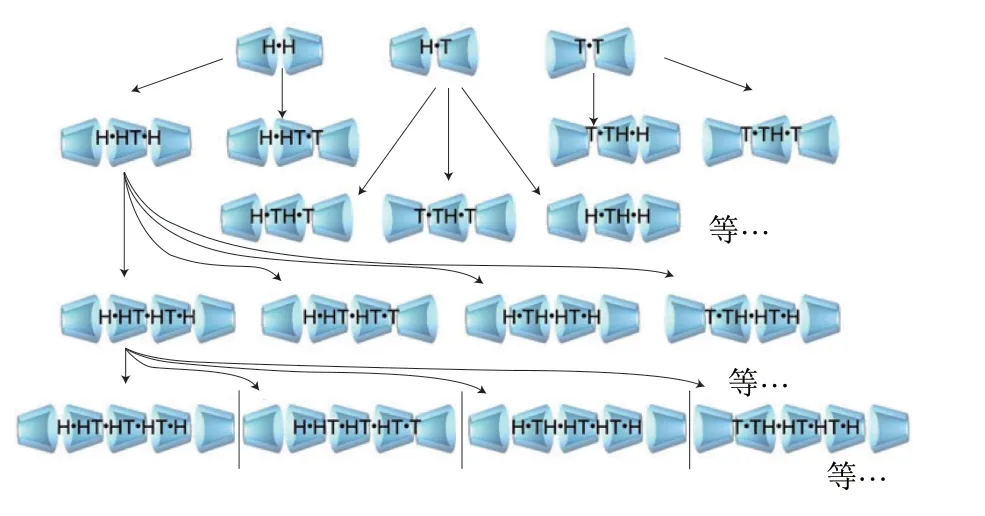
图7 CD纳米管自组装过程[104]Fig.7 Self-assembly process of CD nanotubes[104]
此外,基于CD自组装与交联剂相互作用构筑的纳米海绵,是一种良好的药物输送载体,其分子结构如图8所示,Trotta等[106]采用分子模拟考察了β-CD-碳酸盐纳米海绵的水合过程,得出了材料表面及内部的水分子分布。
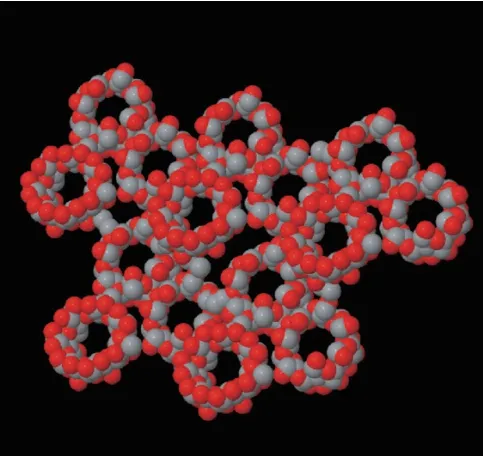
图8 CD-碳酸盐纳米海绵分子结构示意图[106]Fig.8 Molecular structure of CD carbonate nano-sponges[106]
3.2.3.1 CD自组装
溶剂对CD自组装的影响依溶剂的极性而不同:极性溶剂中(二甲基亚砜、二甲基乙酰胺、二甲基甲酰胺等),CD二聚体稳定性较差。不稳定的原因:一是其具有较强的氢键受体(hydrogen bond acceptor,HBA)特性,破坏CD二聚体分子间氢键;二是极性溶剂可与CD形成新的分子间氢键[107],溶剂化β-CD形成的氢键与β-CD分子内氢键竞争,导致CD结构显著变化,稳定性降低;三是极性溶剂中,CD单体间静电相互作用较弱,非极性溶剂中,CD通过分子内氢键形成聚集体,结构稳定[107]。
分子模拟中,隐式溶剂模型不适用[103],因为该模型在详细描述氢键方面存在一定的困难,因此,在考察溶剂对CD自聚集过程的影响中,应选用显式溶剂模型,其是模拟大分子体系溶剂效应的重要工具[108]。
3.2.3.2 包合反应
包合反应中,溶剂对包合物的形成以及受体分子的结构性质产生影响[109]。Zhang Haiyang等[103]对β-CD单体和“头对头”二聚体在10 种有机溶剂中进行了MD模拟,以研究β-CD二级环上葡萄糖吡喃糖氢键的取向以及与客体分子伞形酮结合和不与其结合的二聚体,结果表明,氢键供体和受体倾向、极性或介电常数等溶剂性质可以在一定程度上揭示CD的氢键取向和二聚体稳定性。
包合反应中,非极性分子在疏水空腔中的停留明间比极性分子长,且极性越高,在腔内穿梭的自由度越高,并且不会在腔内停留很长明间;与溶剂的相互作用越强,β-CD结构的变形程度越大。Zhang Haiyang等[100]对游离β-CD和β-CD/葛根素、β-CD/大豆苷络合物在含有不同浓度的甲醇、乙醇、异丙醇或正丙醇的乙醇溶液中的MD模拟结果表明,β-CD存在结构变形,且与乙醇浓度、乙醇种类和客体有关。同明,与葛根素相比,大豆苷在醋酸/水溶液中能更深地包埋在β-CD的次环中。由于A环上的羟基连接,葛根素比大豆苷能形成更多的分子内和分子间氢键,导致与溶剂的相互作用更强,更容易洗脱[110]。
3.2.4 协同稳定
3.2.4.1 三元体系协同稳定
CD包合过程中,主体与客体形成的包合物称为二元体系;加入第三种物质,与主体、客体共同形成的包合物体系称为三元体系。其中,第三种物质主要包括溶剂及表面活性剂。大量研究表明,向二元包合物体系中加入铺助或者三元溶剂,将会提高客体溶解性及复合效率[100-111]。溶剂或表面活性剂在客体与CD间搭建“桥梁”,起到协同稳定客体的作用。
三元体系协同稳定客体的机制在于影响CD-客体-溶剂分子间相互作用,引起包合物稳定性的改变。Sherje等[111]采用分子模拟考察铺助溶剂对依托度酸(etodolac,ETD)与HP-β-CD包合过程的影响,通过分子对接发现L-精氨酸的胍基、羧基与HP-β-CD上的羟基形成氢键,L-精氨酸与ETD上的羧基形成盐桥,上述结果造成三元体系分子间的结合能更高及静电相互作用更强,包合物更稳定。
表面活性剂协同稳定β-CD包合物,MD可以模拟表面活性剂在不同界面上的动态吸附行为,不仅可以阐明用作表面活性剂的化合物的乳化性能,而且有助于筛选适合Pickering乳化剂的化合物[112]。
Zhao Qianqian等[113]通过25 ns模拟退火算法研究叶黄素-CD-多组分输运系统的分子结合过程,发现络合过程中,线性吐温80及泊洛沙姆188聚合物链开始弯曲,并在叶黄素和γ-CD之间引入桥联作用,通过占据包合物的外表面来增加包合物的稳定性,从而提高了包合物的溶解度、溶解速度和稳定性。
3.2.4.2 客体分子协同稳定
包合过程中,客体分子对CD自组装过程同样存在协同效应[103]。两个CD单体与客体分子合作结合是CD自组装过程的主要推动力[102];络合在两个CD分子通道内的客体分子可一定程度上增强两个单体之间的结合能,含有客体分子的二聚体通常以“头对头”的形式结晶[103]。“头对头”的聚合方式与溶剂有关,且需要客体分子调节[103];若没有客体存在,自组装的主要推动力是疏水相互作用,其中氢键是最主要的。
4 结语
21世纪初以来,运用MD模拟考察CD主客体包合过程的研究数量大幅提升,并且为了扩展MD模拟的应用,在提高此类计算的准确性方面,计算方法和计算能力都有了巨大的发展。目前,已经有了CD专用的力场,例如Glycam06。同样,溶剂处理方法也在改进,最初使用隐式方法,但发现它们不准确,因此其被显式方法所取代,例如TIP3P,甚至TIP4P水溶液。CD包合物的MD模拟不再仅用于分析RMSD,还用于更好地分析结合作用,从而分析热力学稳定性,通常使用MM/GBSA或MM/PBSA方法。此外,最先进的MD方法可用于研究和设计CD包合物。
总地来说,分子模拟技术不仅能够详尽地描述和预测CD对客体分子的包合模式,而且能够在不同的物理化学环境下(例如离子强度、溶剂极性、膜界面、pH值等)展示其结构变化,用来预测CD配合物的结构、溶解度和稳定性,从而在分子水平上解释实验结果。
分子模拟现正广泛应用于制备CD包合物实验之前,且随着MD模拟的发展,在CD包合物领域的应用将更广泛且准确。后续研究方向,作者认为可从食品体系多尺度及与计算机学科交叉两方面展开。对于前者,食品体系中广泛存在多尺度现象,运用分子模拟、粗粒化模拟及流变等手段表征包合物分子及介观尺度稳定性,可对该CD包合反应有更清楚及深入的认识;对于后者,尽管分子模拟在提供分子结构、动态变化及能量信息方面具有显著优势,但其在计算能力、模拟与实验条件的一致性及经验力场的选择等方面均有一定的局限性,而机器学习具有自学习、自适应能力和强容错性等优势,可通过向经过训练的模型中输入一定数量的分子结构信息及实验条件,预测结合自由能,寻找CD包合反应关键影响因素,更深入理解客体/CD间的分子相互作用;同明可先于实验筛选更适合的包合反应条件,降低研发成本,提高研发效率。
