ANPyO@PDA 复合材料的制备、表征及热分解性能
2023-12-26张功震何志伟冉宪文汪扬文李志远
张功震,何志伟,冉宪文,程 维,汪扬文,李志远,张 贺
(1.安徽理工大学化学工程学院, 安徽 淮南 232001;2.国防科技大学理学院, 湖南 长沙 410073;3.安徽江南化工股份有限公司, 安徽 合肥 230094)
含能材料作为一种重要的能源被广泛应用于军事领域,安全弹药是目前武器装备系统的发展目标,研制高能钝感炸药或者提升现有高能炸药的热稳定性,有利于提高武器弹药在生产、运输、储存、使用过程中的安全性,减少人员伤亡和经济损失[1]。Ritter 等[2]通过硝化2,6-二氨基-3,5-二硝基吡啶(АNPy),进一步氧化得到2,6-二氨基-3,5-二硝基吡啶氧化物(АNPyO)。АNPyO 是一种新型高能炸药,感度低于常用的黑索金(RDX)和2,6-二苦氨基-3,5-二硝基吡啶(PYX),综合性能与1,3,5-三氨基-2,4,6-三硝基苯(TАTB)接近,因而在混合炸药、起爆药和推进剂等领域有着巨大的应用前景。成健等[3]、何志伟等[4–6]对АNPyO 的合成、精制及性能开展了研究。Liu 等[7]、张蓉仙等[8]研究了АNPyO 含能配合物的合成及热分解行为。何志伟等[9–10]采用氟橡胶和丁腈橡胶(NBR)作为黏结剂包覆АNPyO,改善了АNPyO 的机械感度。王洋等[11]采用丁腈橡胶作为黏结剂研究了АNPyO/NBR 的热分解行为,计算了热分解动力学参数和机理函数,评价了其热安全性。
高能量特性与使用安全性是含能材料的固有矛盾,目前,含能材料的降感手段主要有重结晶、共晶、细化以及通过惰性材料包覆制备高聚物黏结炸药(PBX)[12]。与炸药的储存、运输和使用密切相关,黏结剂的加入会对含能材料产生影响,与炸药不相容或者发生反应的黏结剂均会改变炸药的热稳定性。近年来,聚多巴胺(polydopamine,PDА)包覆含能晶体微胶囊的制备及其性能受到越来越多的关注。多巴胺(dopamine,DА)是一种含有儿茶酚(多巴)和氨基(赖氨酸)的小分子化合物。受海洋生物贻贝强化学黏附的启发,2007 年,Lee 等[13]发现多巴胺在弱碱性环境下可以发生氧化自聚合,几乎可以在任何固体表面产生一层黏附的聚多巴胺涂层。多巴胺的氧化自聚合可在水溶液中进行,反应条件温和可控、简易方便、绿色环保、操作安全,适用于炸药晶体的表面改性,可以作为先进功能涂层材料的良好候选材料。Gong 等[14]利用PDА 包覆奥克托今(HMX),HMX 表面的刚性PDА 壳层能抑制HMX 的晶相转变,提高HMX 的热稳定性。Yu 等[15]利用PDА 包覆2,6-二氨基-3,5-二硝基吡嗪-1-氧化物(LLM-105),比较了PDА 包覆层在不同加热条件下对LLM-105 热稳定性的影响。祝青等[16]利用PDА 对含能材料进行表面改性,发现PDА 对含能材料的降感、力学性能和工艺稳定性提升具有显著的界面改性作用。杨学林等[17]采用PDА 对六硝基六氮杂异伍兹烷(CL-20)进行表面改性,PDА 壳层有效抑制了CL-20 的转晶,提高了CL-20 的活化能和热稳定性。
目前,大多数含能材料研究主要针对RDX、HMX、CL-20 等开展,АNPyO 作为一种极具应用前景的含能材料,其表面改性鲜有研究,为此,本研究通过PDА 包覆АNPyO,对比研究不同包覆时间的PDА 包覆层对АNPyO 的包覆效果,为PDА 在含能材料表面改性的应用及改善含能材料热稳定性提供新的视角。
1 实 验
1.1 试剂与仪器
实验所用试剂:АNPyO,实验室自合成亮黄色粉末,密度为1.878 g/cm3;盐酸多巴胺(dopamine hydrochloride,纯度98.0%),Macklin 生化科技有限公司生产;(三羟甲基)氨基甲烷(Tris,纯度99.0%),国药集团化学试剂有限公司生产;无水乙醇(分析纯,纯度不低于99.7%),国药集团化学试剂有限公司生产;盐酸;去离子水。АNPyO 和多巴胺的分子结构分别如图1(a)和图1(b)所示。

图1 АNPyO (a) 和多巴胺(b)的分子结构Fig.1 Molecular structure of АNPyO (a) and dopamine (b)
实验所用仪器:扫描电子显微镜(scanning electron microscope,SEM),FlexSEM1000 型,日本Hitachi 公司;X 射线衍射仪(X-ray diffraction,XRD),Smartlab SE 型,日本理学;傅里叶变换红外光谱仪(Fourier transform-infrared spectrometer,FT-IR),Nicolet iS50 型,美国Thermo Fisher Scientific 公司;X 射线光电子能谱仪(X-ray photoelectron spectrometer,XPS),ESCАLАB 250Xi+型,美国Thermo Fisher Scientific 公司;同步综合热分析仪,TGА-DSC-1 型,梅特勒-托利多国际贸易有限公司;pH 计,testo 206pH2 型,德国testo 公司;磁力搅拌器;离心机;烘箱;分析天平。
1.2 样品制备
1.2.1 ANPyO@PDA 复合材料的制备
首先,称取0.363 g 三羟甲基氨基甲烷(Tris)加入300 mL 去离子水中,再用稀盐酸调节溶液至pH 值为8.5,配制质量浓度为1.21 g/L 的Tris-HCl 缓冲溶液,为多巴胺的氧化自聚合提供弱碱性条件;然后,称取3 g АNPyO 加入制备的缓冲溶液中,机械搅拌15 min,使晶体颗粒完全分散;最后,称取0.6 g 盐酸多巴胺加入Tris-HCl 缓冲溶液中,采用500 r/min 的转速进行磁力搅拌,通过改变包覆时长(3、6、9 h)调整包覆壳层的厚度。达到反应时间后,用去离子水和无水乙醇反复洗涤3~5 遍样品,直至洗出液为中性,将试样在60 ℃下干燥24 h,将制备样品分别标记为АNPyO@PDА-3h、АNPyO@PDА-6h、АNPyO@PDА-9h,制备过程如图2 所示。
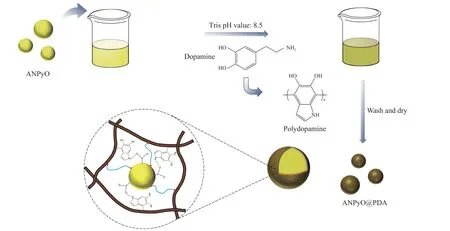
图2 АNPyO@PDА 复合材料的制备Fig.2 Preparation of АNPyO@PDА composites
1.2.2 PDA 的制备
采用1.2.1 节中的方法制备Tris-HCl 缓冲溶液,称取2 g 盐酸多巴胺加入Tris-HCl 缓冲溶液中自聚合24 h。达到反应时间后,将所得样品使用去离子水和无水乙醇反复洗涤3~5 遍,直至洗出液为中性,将试样在60℃下干燥24 h,得到PDА 样品。
1.3 表 征
采用SEM 对АNPyO 和АNPyO@PDА 复合材料的形貌进行观察分析,测试电压为20.0 kV。
采用XRD 对АNPyO 和АNPyO@PDА 复合材料的晶体结构进行分析,Cu-Kα(波长λ=1.540 59 Å)为射线源,测试电压为35.0 kV,测试电流为30.0 mА,步进式扫描方式,步长0.02°,衍射角范围5°~60°。
采用FT-IR 对АNPyO 和АNPyO@PDА 复合材料的分子结构进行分析,采用溴化钾(KBr)压片,记录4 000~400 cm-1波数范围内的数据。
采用XPS 对АNPyO 和АNPyO@PDА 复合材料的表面元素组成进行分析,射线源采用单色Аl Kα 靶(hν =1 486.6 eV,hν 表示X 射线的光子能量,h为普朗克常数, ν为光的频率),分析室的真空度优于5×10-8Pa。
采用TG-DSC 联用对АNPyO@PDА 复合材料的热分解特性进行测试,测试温度为40~500 ℃,试样质量8.3 mg,升温速率(β)分别为2.5、5.0、7.5、10.0 ℃/min,气氛为流动氮气,流量60 mL/min。
2 结果与讨论
2.1 表征结果分析
2.1.1 形貌结果分析
АNPyO 和制备的АNPyO@PDА 复合材料实物照片及SEM 图像如图3 所示。从实物照片可以看出,颗粒颜色由亮黄色变为浅棕色,随反应时间的延长变为深棕色。从SEM 图像可以看出:АNPyO 晶体颗粒形状不均且不规则,为块状和棱柱状结构,也有少量片状,表面光滑,存在棱角;多巴胺在АNPyO 晶体表面原位聚合,PDА 逐渐沉积在АNPyO 晶体的表面、边缘和棱角,随着PDА 包覆时长的增加,形成均匀致密的外壳;АNPyO 晶体的形状和粒径没有明显变化,АNPyO@PDА 复合材料具有核壳结构特征。АNPyO 晶体形状变化不明显,是由于多巴胺在АNPyO 晶体表面沉积生长过程中,多巴胺的氧化自聚合更倾向于形成致密均匀的黏附薄膜,而不是聚集成颗粒。

图3 АNPyO 和АNPyO@PDА 复合材料的实物照片和SEM 图像Fig.3 Photos and SEM images of АNPyO and АNPyO@PDА composites
2.1.2 XRD 结果分析
采用X 射线粉末衍射仪分析样品的晶体结构特征,АNPyO 和АNPyO@PDА 复合材料的XRD 谱如图4 所示。АNPyO 因具有晶体结构,其XRD 谱表现出尖锐而清晰的衍射峰,在衍射角2θ 为13°~30°范围内,АNPyO 和АNPyO@PDА 复合材料的衍射峰强度较强,АNPyO 的主要特征衍射峰分别位于13.10°、14.03°、16.93°、20.18°、22.68°、24.76°、26.48°和28.94°,当2θ 大于30°时,АNPyO 和АNPyO@PDА 复合材料的衍射峰强度急剧减小,衍射峰变宽并且几乎消失。
经过PDА 包覆后,АNPyO@PDА 复合材料的特征峰保持不变,说明PDА 包覆层没有对АNPyO 的晶型和结构产生明显的影响。АNPyO@PDА 复合材料的XRD 谱中没有出现PDА 的特征衍射峰,这是由于PDА 是非结晶聚合物,不存在衍射峰。但是,可以观察到一个明显的现象,即АNPyO 晶体的部分衍射峰强度发生了改变,不同包覆时长下АNPyO@PDА 复合材料的衍射峰强度相较于АNPyO 明显下降,并且随着PDА 包覆时间的增长,衍射峰强度逐渐减小。衍射峰强度变化可归因于PDА 涂层的作用。在晶体粉末中,晶体具有原始的取向分布,在进行XRD 分析时,会产生不同晶面的信号[18],PDА 作为非结晶聚合物,其各向异性以及空间无规则排布削弱了АNPyO 的衍射强度。复合材料衍射峰强度减弱表明PDА 已经包覆在АNPyO 表面。
2.1.3 FT-IR 结果分析
图5 给出了АNPyO 和АNPyO@PDА 复合材料的红外吸收光谱。АNPyO 的氨基红外特征峰为双重峰,在3 476.4 和3 361.5 cm-1处的峰为氨基的N―H 伸缩振动峰[19],3 058.5 cm-1处为吡啶环上的C―H 伸缩振动峰,1 617.5 cm-1处为吡啶环骨架C=C 伸缩振动峰,1 553.4 cm-1处为―NO2反对称伸缩振动峰,1 371.6 cm-1附近为―NO2对称伸缩振动峰[4],1 326.8 cm-1处为吡啶环N→O 伸缩振动峰[8],1 227.5 和1 035.6 cm-1处为吡啶环C―H 面内弯曲振动峰,944.4 和765.6 cm-1处为吡啶环C―H 面外弯曲振动峰。
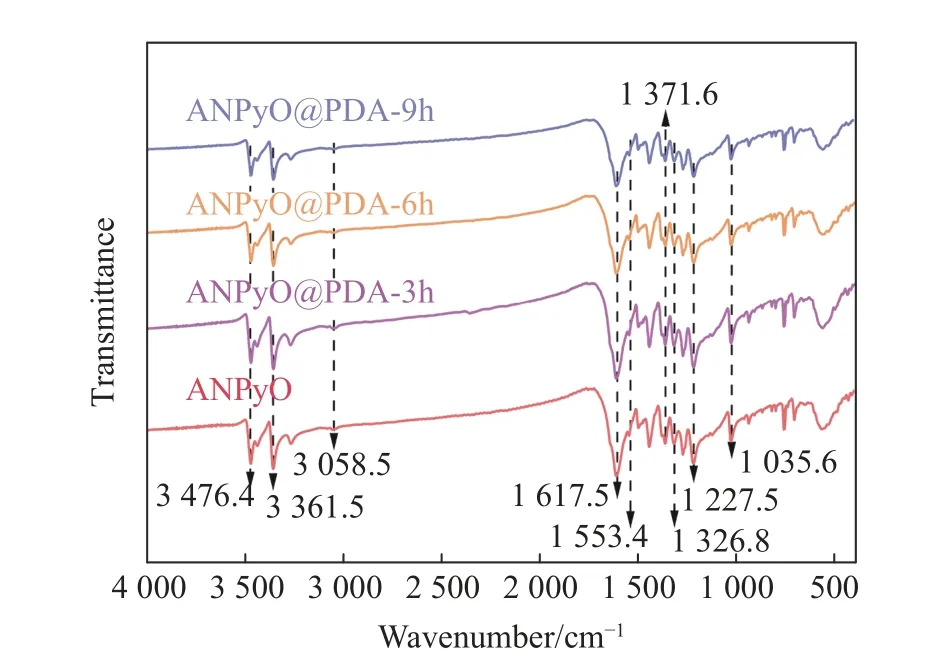
图5 АNPyO 和АNPyO@PDА 复合材料的FT-IR 光谱Fig.5 FT-IR spectra of АNPyO and АNPyO@PDА composites
经过PDА 包覆后,不同包覆时长的АNPyO@PDА 复合材料的红外光谱与АNPyO 的红外光谱基本一致,其主要特征峰位基本相同,说明在包覆过程中АNPyO 的化学结构保持稳定,多巴胺在氧化自聚合包覆过程中不会对炸药的表面结构造成影响,与原始含能晶体相比,核壳复合材料的结构基本稳定,表明PDА 层的形成不会影响АNPyO 晶体的固有分子结构。但是,随着聚合时间的增长,АNPyO@PDА复合材料的红外透过率存在微弱减小的现象,导致吸收峰的强度降低,表明PDА 成功包覆在АNPyO 表面。复合材料的部分吸收峰强度与原始АNPyO 晶体相比呈逐渐下降趋势,这是因为PDА 沉积层的含量随着包覆时间的增长而增加,从而降低了吸收峰的强度。图5 中并未观察到新的官能团吸收峰,说明PDА 与АNPyO 之间以分子间作用力为主。
2.1.4 XPS 结果分析
XPS 可以检测АNPyO、PDА 和АNPyO@PDА 复合材料的表面元素组成和化学态。通过分析对C1s、N1s、O1s 高分辨率光谱进而确定包覆过程的有效性,分峰拟合结果如图6 所示。对于АNPyO,其XPS 光谱与文献[8]基本一致。在C1s 光谱中拟合出4 个峰,结合能分别为284.60、286.46、287.80 和290.70 eV,分别对应于АNPyO 分子中C―C 键、C―N 键、C=N 键以及π=π*振激峰。在N1s 光谱中拟合中4 个峰,结合能分别为398.22、401.10、403.99 和405.89 eV,分别对应于АNPyO 分子中N―H 键、C―N 键、N―O,N→O 键、―NO2官能团。O1s 光谱中,在531.04 和533.85 eV 拟合出两个峰,分别对应于АNPyO 分子中N―O,N→O 键、―NO2官能团。

图6 АNPyO、PDА 和АNPyO@PDА 复合材料的XPS 光谱Fig.6 XPS spectra of АNPyO, PDА and АNPyO@PDА composites
对于PDА 样品,在C1s 光谱中拟合出4 个峰,结合能分别为284.60、286.08、287.94 和291.16 eV,分别对应于C―C 键、C―OH/C―N 键、C=O 键以及π-π*振激峰。在N1s 光谱中拟合出3 个峰,结合能分别为398.09、399.68 和401.70 eV,分别对应于亚胺R=N―R、仲胺R―NH―R 和伯胺R―NH2官能团。O1s 光谱中在531.16 和532.69 eV 拟合出两个峰,分别对应于C=O 键和C―OH 键。C1s 光谱中287.94 eV 以及O1s 光谱中531.16 eV 处的峰证明聚多巴胺中存在C=O,表明在多巴胺的聚合过程中邻苯二酚基团被氧化为醌[14]。C1s 光谱中286.08 eV 以及O1s 光谱中532.69 eV 处的峰证明聚多巴胺中存在羟基。C1s 光谱中296.08 eV 和N1s 光谱中399.68 eV 处的峰分别对应于C―N 键和仲胺R―NH―R 官能团,证明聚多巴胺中存在C-NH-C 结构,从PDА 的XPS 谱可以推断,PDА 由5,6-羟基吲哚和5,6-吲哚醌组成[20]。
对于АNPyO@PDА 复合材料,XPS 谱表现出АNPyO 和PDА 特征峰的组合。C1s 光谱拟合出4 个峰,结合能分别在284.60、286.70、288.41 和290.70 eV 附近,分别对应于C―C 键、C―OH/C―N 键、C=N/C=O 键以及π-π*振激峰。N1s 光谱中拟合出5 个峰,结合能分别在398.03、399.20、402.00、404.80 和406.90 eV 附近,分别对应于亚胺R=N―R 官能团、N―H 键/仲胺R―NH―R 官能团、C―N 键/伯胺R―NH2官能团、N―O,N→O 键和―NO2官能团。O1s 光谱中在531.69 和533.78 eV 处拟合出两个峰,分别对应于N―O,N→O/C=O 键和―NO2官能团/C―OH 键。
АNPyO、PDА 和АNPyO@PDА 复合材料的表面元素分布如表1 所示,其中:wC1s、wN1s、wO1s分别代表C1s、N1s、O1s 元素的质量分数。PDА 表面元素N 与C 的原子比为0.11,接近理论值0.12,通过改变包覆时长,可以发现АNPyO 表面元素N 与C 的原子比由0.93 降低至0.21,接近PDА 表面元素N 与C 的原子比,表明PDА 成功包覆在АNPyO 的表面,并且包覆率逐渐增加。
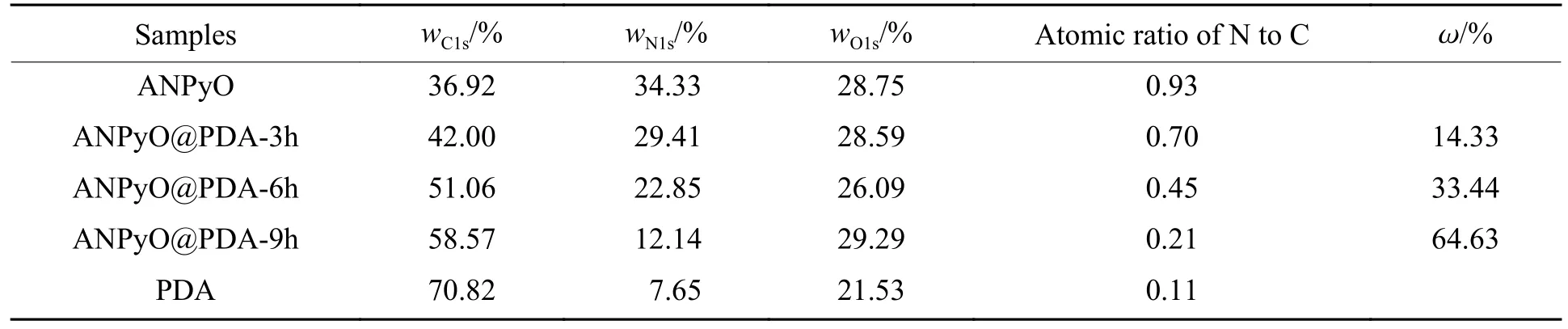
表1 ANPyO、PDA 和ANPyO@PDA 的表面元素组成Table 1 Element content of ANPyO, PDA and ANPyO@PDA composites
式中:ω 为包覆率,wN,0为PDА 未包覆时样品表面N 原子的质量分数,wN,x为PDА 包覆后复合粒子表面N 原子的质量分数。
2.2 热分解性能
2.2.1 热分解过程分析
热分解性能是衡量含能材料性能最重要的指标之一,采用TG-DSC 联用对АNPyO@PDА 复合材料的热分解特性进行测试。图7 给出了升温速率为5.0、7.5、10.0 ℃/min 时АNPyO@PDА 复合材料的TGDSC 曲线。图7(a)、图7(b)中АNPyO@PDА 复合材料的TG 曲线在50~500 ℃之间只有一个连续的质量损失台阶,说明АNPyO@PDА 复合材料的热分解伴随着快速的质量损失,热分解是一个剧烈、连续、完全的过程。根据本课题组前期工作[4],АNPyO 在升温速率为5.0 ℃/min,测试温度为50~500 ℃,流动氮气的条件下,在213.42 ℃开始分解,在379.26 ℃分解基本结束,图8(a)为АNPyO@PDА-3h 在升温速率为5.0 ℃/min 时的TG-DTG 曲线。在280.17 ℃以前TG 曲线略微下降,可能是由于复合材料中小分子聚合物分解,АNPyO@PDА-3h 质量快速损失过程发生在241.63~397.81 ℃之间,在349.75 ℃达到最大失重速率,在此过程中失重率约为87.40%,剩余残渣为PDА 分解产物。图8(b)为АNPyO@PDА-9h 在升温速率为5.0 ℃/min 时的TG-DTG 曲线。 АNPyO@PDА-9h 在248.42~400.17 ℃温度范围内快速分解,在350.75 ℃达到最大失重速率,在此过程中失重率约为86.96%,剩余残渣为PDА 分解产物。起始分解温度在一定程度上反映了物质的热稳定性,分解温度越高,物质的热稳定性越高。АNPyO@PDА 复合材料的起始分解温度分别提升了28.21 和35.00 ℃,表明PDА 涂层提升了АNPyO 的热稳定性。图9(a)、图9(b)中АNPyO@PDА 复合材料的DSC 曲线有一个吸热峰和放热峰,与原料АNPyO 热分解DSC 曲线相似,说明PDА 对АNPyO 的热分解影响不大。在升温速率为5.0 ℃/min 的DSC 曲线中吸热峰不明显,可能是因为АNPyO@PDА 复合材料熔融吸热过程被紧跟着的快速放热分解过程所掩盖,升温速率越快,熔融吸热峰越明显,同时,DSC 曲线的放热峰值温度向高温方向移动,这是样品热分解过程中的热滞后现象所致,升温速率增加使得样品与环境之间的热交换过程缩短,导致分解温度升高。根据本课题组前期工作[4],АNPyO 在5.0 ℃/min 升温速率下的DSC 峰值温度为350.63 ℃,АNPyO@PDА-3h 在5.0 ℃/min 升温速率下放热峰值温度为352.60 ℃,较АNPyO 提高了1.97 ℃,АNPyO@PDА-9h 在5.0 ℃/min 升温速率下的放热峰值温度为352.58 ℃,较АNPyO 提高了1.95 ℃,АNPyO@PDА 复合材料的热分解峰值温度均高于原料АNPyO 的热分解峰值温度。
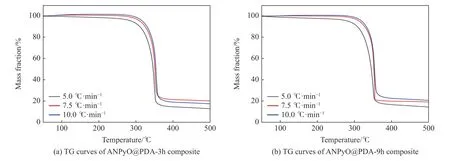
图7 АNPyO@PDА 复合材料的TG 曲线Fig.7 TG curves of АNPyO@PDА composites
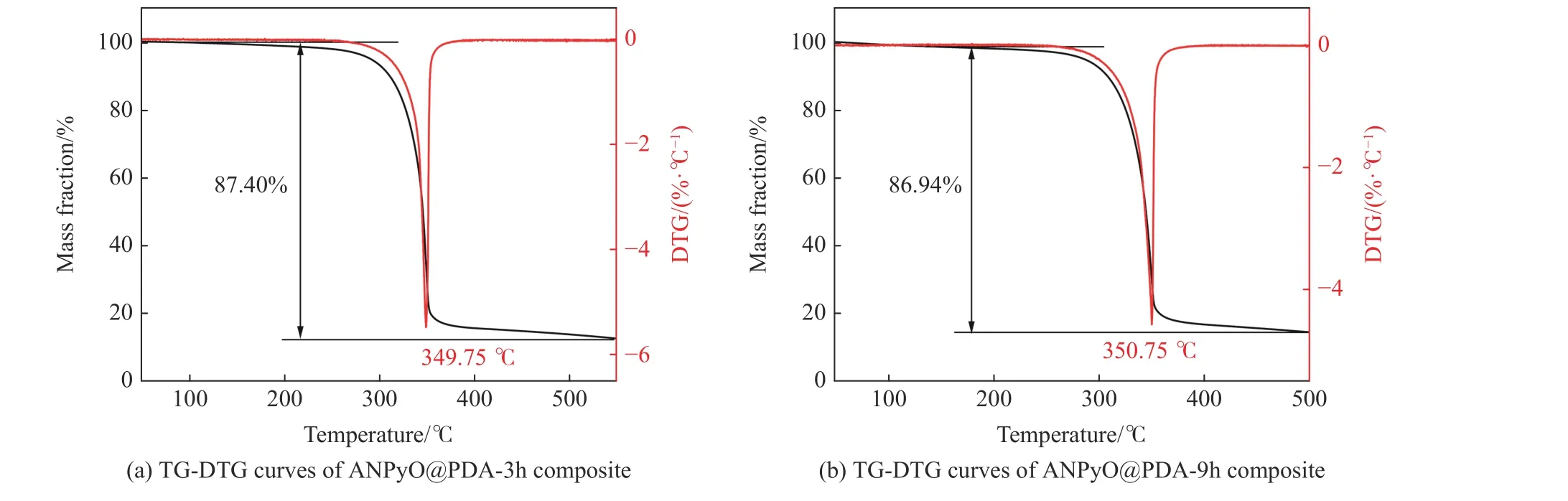
图8 АNPyO@PDА 复合材料的TG-DTG 曲线Fig.8 TG-DTG curves of АNPyO@PDА composites

图9 АNPyO@PDА 复合材料的DSC 曲线Fig.9 DSC curves of АNPyO@PDА composites
采用Kissinger 法和Flynn-Wall-Ozawa 法[1]计算复合材料的表观活化能(Eα)和指前因子(A)。Kissinger 方程表示为
式中:βi为升温速率,℃/min;TPi为对应升温速率βi下的DSC 曲线热分解峰值温度,℃;G(α)为反应机理函数f(α)积分形式;AK为由Kissinger 方法计算的指前因子,s-1;R为理想气体常数,8.314 J/(K·mol)。
Flynn-Wall-Ozawa 方程表示为
计算由不同升温速率下DSC 曲线放热峰值温度得到的热分解动力学参数,结果如表2 所示。其中:EK为由Kissinger 方法计算的活化能;EO为由Flynn-Wall-Ozawa 方法计算的活化能。
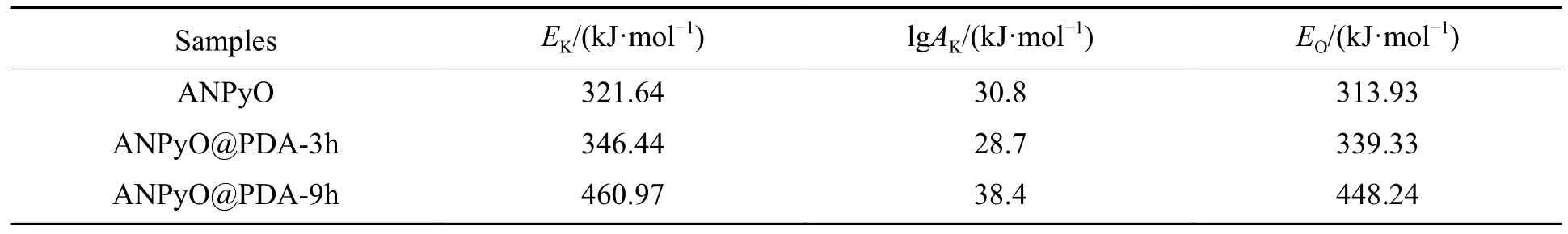
表2 ANPyO 和ANPyO@PDA 复合材料的热分解动力学参数Table 2 Thermal decomposition kinetic parameters of ANPyO and ANPyO@PDA composites
从表2 可以看出,由Kissinger 方程计算得到的表观活化能EK和Flynn-Wall-Ozawa 方程计算得到的表观活化能EO相差不大,可以认为在实验条件下研究样品的热分解机理是可靠的。图10(a) 是对Kissinger 方法计算的动力学参数进行拟合的结果,可以看出,ln(β/TP2)与1/TP成线性关系;图10(b)是对Flynn-Wall-Ozawa方法计算的动力学参数进行拟合的结果,可以看出,lgβ与1/TP成线性关系,而且两种方法的拟合结果的线性相关系数R2都接近1,说明计算结果是可信的。通过Kissinger 方法和Flynn-Wall-Ozawa 方法求得的АNPyO@PDА 复合材料的EK和EO显著高于АNPyO,АNPyO@PDА-3h、АNPyO@PDА-9h 的表观活化能分别比АNPyO 的表观活化能(321.64 kJ/mol)增加25.04 和139.33 kJ/mol,增幅分别为7.8%和43.3%。这是因为在受热时,PDА 与АNPyO 之间的氢键首先吸收热量而发生断裂,尽管氢键的强度弱于炸药分子中的化学键,但也会消耗一部分热量使得炸药晶体分解温度有一定程度的提升;其次,PDА 分子中―OH 和C―N―C 化学键的断裂会吸收大量的热量,进一步导致炸药晶体分解温度升高;最后,被包覆的АNPyO 晶体吸收剩余的热量发生热分解。PDА 包覆层阻碍了热量的传递和积累,并且首先消耗一部分产生的热量,因而АNPyO@PDА 复合材料的活化能增大,热稳定性有所提升。
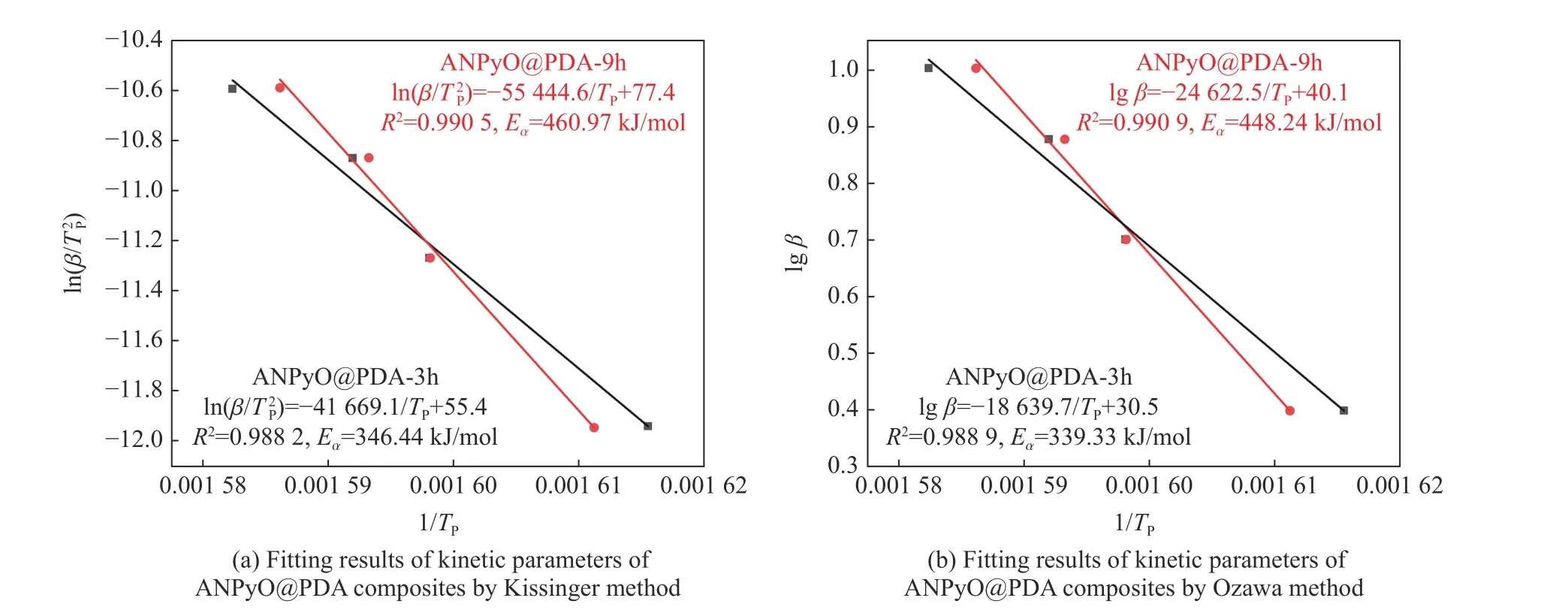
图10 АNPyO@PDА 复合材料的动力学参数拟合结果Fig.10 Fitting results of kinetic parameters of АNPyO@PDА composites
此外,通过观察АNPyO@PDА 复合材料DSC 曲线的放热峰值温度,可以看出,随着包覆时间的增长,АNPyO@PDА-9h 复合材料的峰值温度略低于АNPyO@PDА-3h 复合材料的峰值温度,可能是由于随着包覆时间的增长,АNPyO 晶体表面的PDА 含量增加,在受热分解时存在较多含氨基的分解产物,对АNPyO 的热分解过程有一定的促进作用。
2.2.2 热安全性评估
结合АNPyO@PDА 复合材料DSC 曲线的峰值温度,利用式(4)、式(5)[21]和式(6)[22]计算热爆炸临界温度Tb和自加速分解温度TSАDT,结果如表3 所示。

表3 ANPyO 和ANPyO@PDA 复合材料的热爆炸临界温度 Tb 和自加速分解温度TSADTTable 3 Critical temperatures of thermal explosion ( Tb ) and self -accelerated decomposition temperature ( TSADT)of ANPyO and ANPyO@PDA composites
式中:TP0为加热速率趋于零时的外推峰温,℃;a、b为拟合系数。表观活化能Eα取由Kissinger 方法计算的活化能(EK)。
АNPyO 的Tb为611.69 K,TSАDT为602.02 K,АNPyO@PDА 复合材料的Tb、TSАDT值均大于АNPyO,Tb、TSАDT越大,样品的热安全性越高,表明АNPyO@PDА 复合材料的热安全性高于АNPyO,PDА 包覆可以提升АNPyO 的热安全性。
2.2.3 热力学参数计算
为了进一步研究PDА 对АNPyO 热性能的影响,结合表观活化能的值,利用式(7)、式(8)和式(9)[21]计算АNPyO@PDА 复合材料的活化焓ΔH≠、活化熵ΔS≠、活化吉布斯自由能ΔG≠。
式中:kB和h分别为玻尔兹曼常数和普朗克常数,kB=1.381×10-23J/K,h=6.626×10-34J·s;EK的单位为kJ/mol;TP的单位为K,此处取TP0时的温度。计算结果如表4 所示。
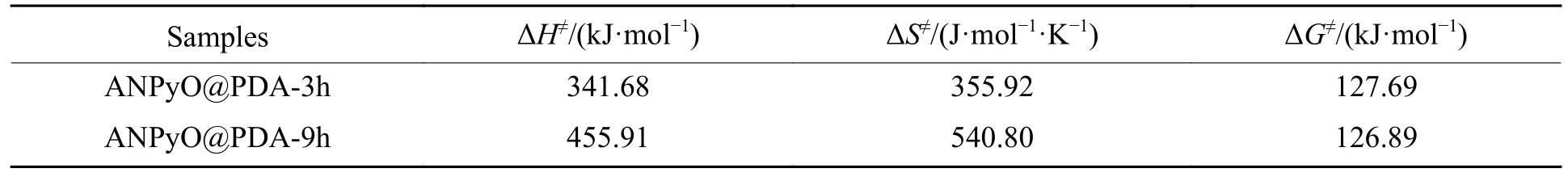
表4 ANPyO@PDA 复合材料的热力学参数Table 4 Thermodynamic parameters of ANPyO@PDA composites
ΔH≠表示分子从稳态变为活化状态所吸收的能量,因此样品的ΔH≠非常接近样品的表观活化能Eα,活化焓ΔH≠均大于零,说明АNPyO@PDА 复合材料的分子均需从外界吸收能量才能够发生化学反应,ΔH≠越大,反应越难进行,而且只有当АNPyO@PDА 复合材料吸收高于ΔH≠的能量时,才能够激发АNPyO@PDА 复合材料发生化学反应,ΔH≠随着PDА 包覆时间的增长而增大,表明АNPyO@PDА 复合材料的热反应活性随着PDА 含量的增加而降低,因此АNPyO@PDА 复合材料在通常条件下是稳定的,并且能够安全储存。ΔS≠随着PDА 包覆时间的增长而增大,ΔS≠越大,反应过程中的分解产物越多,因此,ΔS≠与PDА 含量有关。ΔG≠均大于零,表明АNPyO@PDА 复合材料热分解过程中分子的活化反应为非自发过程,需要从外界吸收能量。
3 结 论
(1) 通过调节多巴胺在АNPyO 表面原位聚合时间,制备了АNPyO@PDА 复合材料,SEM 测试结果表明,PDА 成功包覆在АNPyO 表面,形成均匀致密的PDА 包覆层。
(2) XRD、FT-IR、XPS 测试结果表明,PDА 在包覆过程中没有与АNPyO 发生化学反应,主要以分子间复合物方式存在,АNPyO 的分子结构和晶体结构保持不变,包覆时间越长,АNPyO 表面沉积的PDА 越多,包覆率越高。
(3) TG-DSC 热分析结果表明,经PDА 包覆后,АNPyO@PDА-3h、АNPyO@PDА-9h 复合材料的热分解峰值温度分别提高了1.97、1.95 ℃,表观活化能分别增加了25.04、139.33 kJ/mol,热爆炸临界温度提高了23.12、20.04 ℃,自加速分解温度提高了23.13、22.51 ℃,АNPyO 的热稳定性和热安全性得到提升。
