高压固相拓扑聚合合成纳米碳材料
2023-12-26费云帆郑海燕
费云帆,李 阔,郑海燕
(北京高压科学研究中心, 北京 100193)
碳是自然界中最重要的元素之一,具有sp、sp2和sp3多种杂化方式,可以通过不同的成键方式形成结构多样的碳材料。例如,sp3碳构成的金刚石,sp2碳形成的富勒烯、碳纳米管、石墨烯纳米带,以及sp和sp2碳共同组成的石墨炔等。碳材料结构的多样性导致其功能的多样性。碳材料在硬度、光学特性、耐热性、导电性等方面具有优异性能,拥有广阔的应用前景。通过对碳材料结构的控制可以实现对性质的调控。因此,开发具有新结构的碳材料和实现对碳材料原子级别的精准可控合成是拓展碳材料性质及应用的重要前提。目前碳材料的合成主要有“自上而下”和“自下而上”两种策略。自上而下合成主要是指从石墨烯、碳纳米管等出发,通过电子束和等离子刻蚀[1–3]以及化学氧化[4]等方法制备碳材料。自上而下策略可以方便快捷地制备出尺寸大致可控的碳材料,但难以实现原子级别的精准调控。自下而上合成则是从小分子出发,主要通过溶液合成和表面合成的方法制备碳材料。自下而上策略通过精确设计反应前体分子实现纳米碳材料的精准、可预测合成[5–8]。然而,该策略目前仍存在明显的不足[9]:溶液合成存在溶解度和反应性产生的反应限制和副反应等问题,制备过程中出现副反应等;表面合成存在反应条件苛刻和规模化制备困难等问题。高压合成作为一种全新的固相合成手段,可以克服反应溶解性以及反应类型有限等问题,是实现碳材料精准合成的另一种重要途径。
压力(压强)是与温度同样重要的热力学参数。提高压力会影响物质的状态和结构,使物质表现出与常压下不同的物理、化学行为。石墨可以在高温高压下通过石墨-金刚石共格界面转变为金刚石[10–15],也可以在室温高压下结合剪切力的作用转变为金刚石[16–20];冷压石墨可以得到M碳[21]、bct-C4[22]、W碳[23]等超硬碳相。C60在27 GPa、1173~1273K下可以形成透明的、几乎完全sp3杂化的无定形碳,该无定形碳具有极高的硬度、弹性模量、导热系数以及可调谐的带隙[24];面心立方C60在30 GPa、1 600 K下可以形成次晶金刚石[25]。对于有机小分子而言,高压使分子间距减小,分子间相互作用增强,电子结构发生变化,原本稳定的结构变得不再稳定,因而,可以引发常压下难以发生的化学反应,生成高压下稳定的化合物。这样的反应往往是不可逆的,意味着高压产物一般可以在常压下稳定存在,使得新型碳材料的高压合成具有实际意义[26]。
在高压科学发展的初期,由于高压技术和表征技术的不足以及对极端条件下化学反应认识的局限,碳基分子的高压行为研究主要集中于相变以及产物的初步表征,没有涉及特殊结构的可控合成。例如,早期的研究认为:苯在高压下发生多次相变[27–32],可以在21 GPa 以上聚合生成含有sp2和sp3碳的无序碳氢化合物[33–35];在光催化下,苯聚合反应的压力可以降至16 GPa,产物仍是非晶态的碳氢化合物[36]。直到2015 年,Fitzgibbons 等[37]在室温下将苯缓慢压缩至20 GPa,得到了一维sp3类金刚石纳米线,开启了利用高压自下而上合成碳材料的大门。
在高压下,通常压力成为反应的主要驱动力,化学反应向体积减小的方向进行。当压力作用于不饱和分子/离子时,具有不饱和键的分子/离子往往会发生聚合反应形成高密度的饱和化合物,称为压力诱导聚合。压力诱导聚合多为固相反应,分子的扩散受到抑制,发生反应时常遵循近邻优先的拓扑化学原则,即反应路径和产物结构高度依赖于反应物的晶体结构、分子空间距离以及相对取向等[38]。由于物质在高压下独特的反应性和高压固相聚合的拓扑特性,压力诱导聚合目前已经成为开发新型碳材料的重要手段。
本文主要介绍含不饱和碳的化合物通过压力诱导聚合合成多种碳材料的研究进展,包括聚烯烃、聚炔化合物、金刚石基纳米线、碳纳米带、石墨烷以及高电荷离子型聚合物,着重介绍利用谱学、晶体学、化学等多种表征方法研究高压反应的过程及成果,并简要介绍高压下有机化学反应规律和反应调控方面的一些认识。
1 聚烯烃
乙烯是最简单的含双键的有机分子,是合成世界上产量最大的聚合物—聚乙烯的起始单体。Wieldraaijer 等[39]用量热法在2.5 GPa、330 K下观察到乙烯的聚合反应。Chelazzi 等[40]发现室温下乙烯在3 GPa 以上聚合生成聚乙烯。反应动力学研究和回收产物的光谱分析表明,不同压力下乙烯分子的间距和相对取向略有不同,从而形成不同的聚合路径。在3.6 GPa 时,固体乙烯的分子排列有利于聚合物沿着a轴线性生长,最终得到高密度结晶聚乙烯;而在5.4 GPa 时,乙烯的各向异性压缩使得分子间距和取向不利于沿a轴的选择性聚合(图1),即反应选择性降低,容易产生支链或构象缺陷,最终导致无定形部分增加,形成较低密度的聚合物。另外,在激光(波长λ≤460 nm)的照射下,乙烯的聚合压力可以降低至0.7 GPa,仍生成高结晶度的聚乙烯[41]。
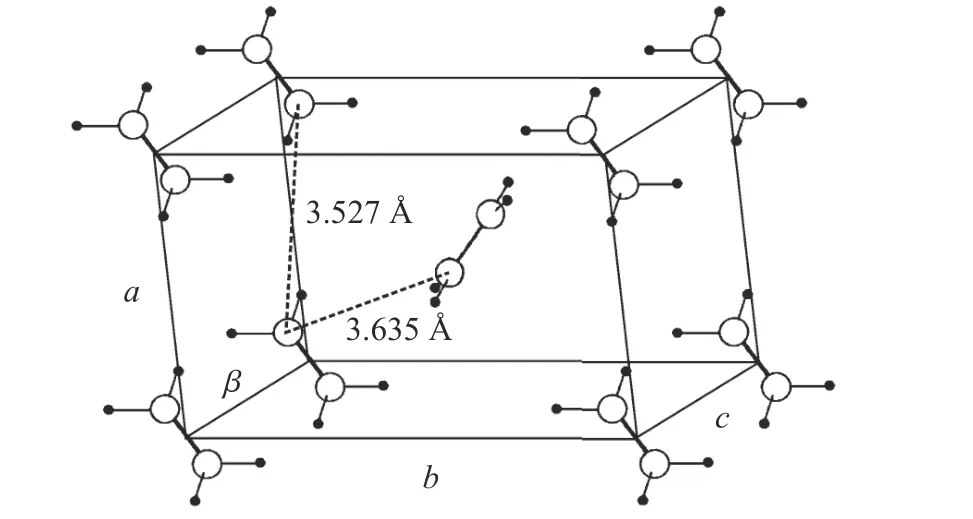
图1 在0.2 GPa、99 K 时乙烯Ⅰ相(P21/n, C 25h , Z = 2)的晶体结构[40](升压产生的各向异性压缩(沿b 轴减小16.7%,沿a 和c 轴分别减小约2%)使沿a 轴的最近邻分子的碳原子间距(3.527 Å)与位于晶胞顶点和中心的分子之间的碳原子间距(3.635 Å)相当)Fig.1 Crystal structure of ethylene in phase Ⅰ (P21/n, C 25h ,Z = 2) at 0.2 GPa and 99 K[40] (This anisotropic compressibility generated by increasing pressure (the cell decreases by 16.7%along the b axis and about 2% along the a and c axis respectively) makes the distances between C atoms of the nearest neighbor molecules located along the a axis (3.527 Å)comparable to those between the molecules sitting on the vertex and at the center of the cell (3.635 Å).)
丁二烯作为最简单的线性共轭烯烃,在环境条件下主要发生二聚反应,生成1,2-二乙烯基环丁烷、1,5-环辛二烯、4-乙烯基环己烯等二聚产物[42]。在液相或高温溶液中使用催化剂和自由基引发剂时,丁二烯可以聚合生成顺式和反式聚丁二烯[43]。丁二烯的高压反应研究发现,由于高压对分子的几何约束,其在0.7 GPa 以上可以发生二聚反应,选择性地生成单一的二聚产物—乙烯基环己烯[44–45]。另外,如果在0.8 GPa 下采用488 nm 的激光照射,二聚反应被完全抑制,丁二烯聚合生成纯反式聚丁二烯[45]。这是因为488 nm 激光的辐照导致丁二烯发生双光子跃迁至S1(21Аg)激发态[45–48],碳碳双键的键长增大[49–50],旋转势垒降低,使末端–CH2基团几乎可以自由旋转,有利于聚合生成反式聚丁二烯,从而表现出高压下光辅助反应的选择性。值得注意的是,光辅助反应的引发依赖于入射波长,即存在引发反应的阈值波长。例如,514.5 nm 的激光无法引发丁二烯的选择性聚合反应,此时丁二烯主要发生纯压力诱导的二聚反应。
2 聚炔化合物
聚乙炔是一类由单双键交替组成的共轭高分子聚合物,经掺杂后具有非常高的导电性[51–52],在电池、半导体材料等方面拥有广阔的应用前景。除掺杂外,利用体积更大或电负性更强的取代基取代聚合物链上的氢原子也可以提高材料的导电性。聚乙炔通常是乙炔在Ziegler-Natta 型催化剂的作用下聚合生成的。高压不仅可以有效地诱导三键发生聚合反应,还能避免取代基引起的催化剂中毒效应,使得在常压下很难聚合的取代炔烃发生聚合。例如,C2I2在0.3 GPa 时由四方相P42/n转变为正交相Cmca,碘原子与相邻碳碳三键之间的供体-受体相互作用明显减弱;在4.0 GPa 以上,C2I2开始聚合,最终形成含大量碳碘(C―I)键的芳香结构或共轭双键结构化合物[53],聚合产物的导电性明显提高。
聚丁炔二酸(acetylenedicarboxylic acid,АDCА)是锂离子电池电极的理想材料,理论容量为470 mАh/g。丁炔二酸可由羧基取代乙炔上的氢得到。由于羰基的电子和空间效应以及酸性质子的存在可使Ziegler-Natta 型或其他过渡金属催化剂失活[54–58],目前只有MoCl5-Ph4Sn 催化剂能使АDCА 聚合,但产率仅为13%,且聚合分子量低[59]。γ 射线也可引发АDCА 聚合,但产率仅为5.5%,并且过度辐照还会使АDCА 降解[60]。高压可在不使用催化剂的情况下引发取代炔的聚合,是生成导电碳骨架的有效手段。原位飞行时间中子衍射表明,АDCА 在4 GPa 时从相Ⅰ(P21/n)转变为相Ⅱ(Pn),在8 GPa 时发生聚合反应生成晶态聚合产物。通过分析临界压力(8 GPa)下的晶体结构和16 GPa 时的中子衍射结果,并结合分子动力学(molecular dynamics,MD)模拟结果(图2),可以发现碳碳三键沿b-c方向聚合。通过Rietveld 精修确定了聚丁炔二酸的晶体结构是沿b轴平行堆积的反式聚乙炔链。碳链上的每个碳原子都连着一个羧基,聚合物主链由链间氢键连接,形成聚合物网络[61]。产物的中子对分布函数也证实了这一结构。此外,凝胶渗透色谱的结果表明聚合压力越高,产物的聚合度越高。电化学研究结果显示,АDCА 具有较高的比容量、优异的循环稳定性和良好的倍率性能,因此,АDCА 可作为锂离子电池的电极材料。
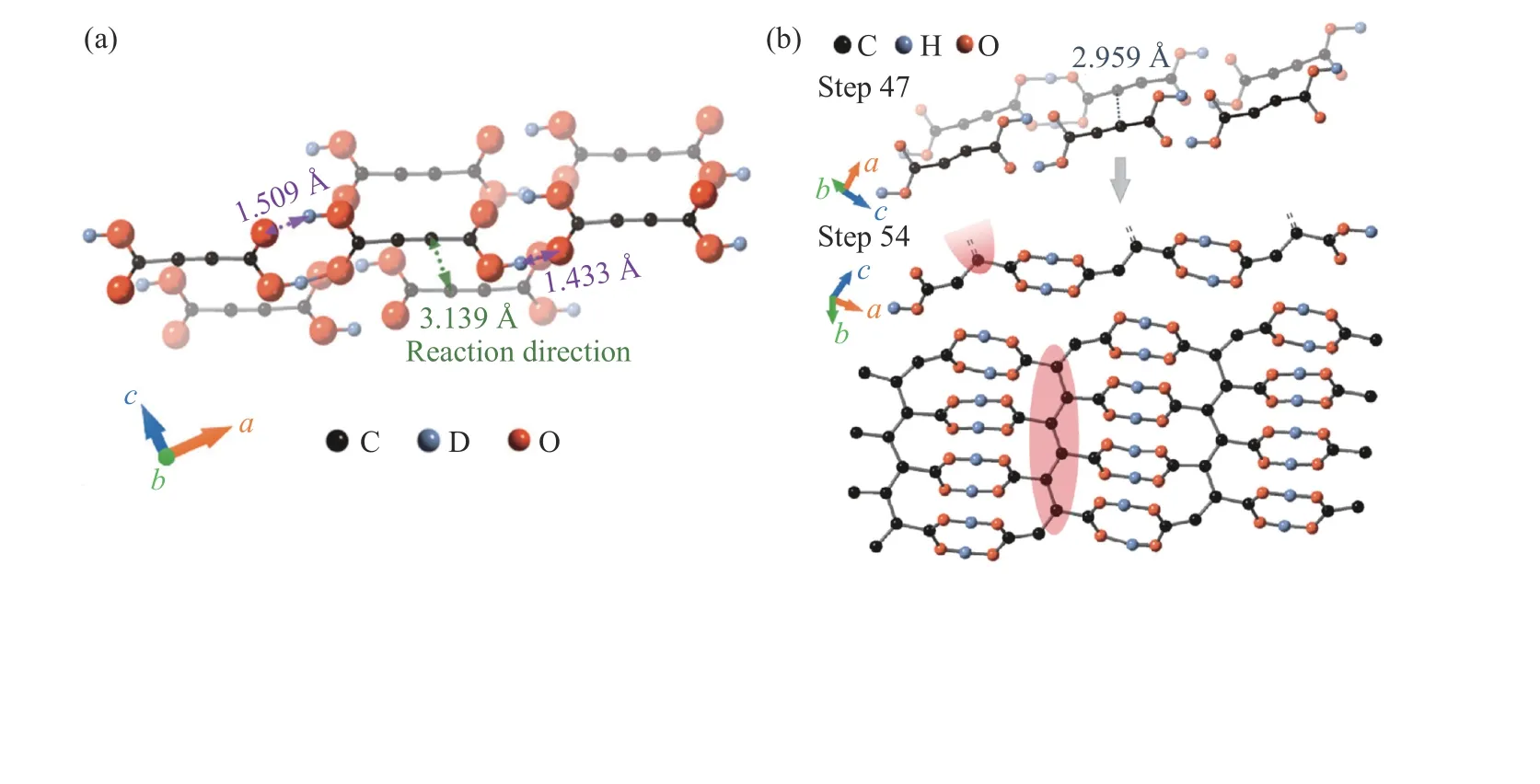
图2 (a) 氘代丁炔二酸(deuterated АDCА,АDCА-d2)在8 GPa 时的晶体结构[61](绿色和紫色箭头分别代表C···C 间距和O···D 间距);(b) 300 K、9 GPa 时АDCА 的分子动力学模拟结果[61];(c) 从16 GPa 回收的产物的晶体结构[61]Fig.2 (a) Crystal structure of АDCА-d2 at 8 GPa[61] ( The green and purple arrows indicate the C···C and O···D distances respectively); (b) molecular dynamics simulation of АDCА at 300 K and 9 GPa[61];(c) crystal structure of the product recovered from 16 GPa[61]
3 金刚石基纳米线
金刚石基纳米线是一类由sp3碳组成的一维纳米材料,拥有高强度、高硬度、高韧性、高杨氏模量、高热导率以及宽带隙等优点,在高强度材料、传感器件以及光电器件等领域具有潜在的应用价值。早在2001 年,Stojkovic 等[62]已经预测出一维sp3C-H 聚合物的存在,命名为tube (3,0)(结构见图3(a))。之后,在2011 年和2014 年,polymer-Ⅰ[63]和polytwistane[64–65]这两种一维sp3碳纳米线结构也被预测出来(见图3(b)和图3(c)))。2015 年,Fitzgibbons 等[37]在苯的缓慢压缩实验中首次得到一维sp3碳纳米线,但该纳米线内部存在诸多缺陷,结构复杂。后来,Li 等[66]通过对苯晶体缓慢施加单轴压力,得到了一维sp3碳纳米线晶体。X 射线衍射(X-ray diffraction, XRD)表明,该sp3碳纳米线在垂直于单轴压力的平面上具有二维有序的伪六方堆积,但在纳米线的轴向上是无序的,即线内结构无序。他们构建并优化了包括tube (3,0)、polymer-Ⅰ和polytwistane 在内的多种纳米线模型,在对比实验和理论模型的特定晶面间距后发现,tube (3,0)和polytwistane 模型的计算值(热校正之后)与实验值吻合得很好。为了进一步研究苯纳米线的线内及线间结构,Maryasin 等[67]利用固体核磁共振(nuclear magnetic resonance, NMR)发现苯纳米线中可能存在polytwistane 的结构片段。Duan 等[68]进一步从富含13C 的苯纳米线出发,利用固体核磁共振和红外光谱进行综合分析,指出高压下苯形成的金刚石基纳米线是多种结构的混合物,其中:约有20%~45%的苯转化为长度至少为2.5 nm、饱和度(每个芳环上sp3碳的数量)为6 的金刚石基纳米线,且产物具有多个成键位点(而非单一位点成键);另外,约1/3 的苯形成了具有孤立双键、饱和度为4 的纳米线;产物中不存在饱和度为2 的纳米线。Wang 等[69]通过密度泛函理论(density functional theory, DFT)计算得到了几种纳米线模型13C-NMR 的化学位移、化学屏蔽张量和化学位移各向异性,并与实验结果进行对比发现,苯纳米线是饱和度为6 和4 的纳米线组合,并且在苯纳米线形成过程中存在[4+2]环加成反应途径。Juhl 等[70]的高分辨透射电镜结果证明了苯纳米线的线性结构和线间的六方对称性,苯纳米线主要由sp3碳(质量分数为73%)构成,并且在轴向上具有纳米级的平移有序性。为了进一步认识苯在高压下的聚合反应路径和产物结构,Xu 等[71]列举了所有饱和度为6 的苯纳米线,发现了至少50 种不同的稳定结构,其中最稳定的15 种结构包括tube (3,0)、polymer-Ⅰ和polytwistane。Chen 等[72]发现苯在高压下存在多种反应路径,根据苯可能的聚合方向提出了[4+2] Diels-Аlder 聚合、[2+2]聚合和对位聚合3 种反应途径,并对比了所有饱和度为2 和4 以及部分饱和度为6 的纳米线的能量,研究结果表明,饱和度为6 的纳米线的稳定性最高,且高于苯分子。总之,目前的理论与实验结果均表明,苯在高压下的聚合产物具有多种金刚石基纳米线结构,且反应路径较为复杂。其主要原因为苯分子之间较弱的π···π 相互作用以及6 个碳原子的无差别反应性。

图3 一维sp3 C-H 纳米线的3 种预测结构[72]Fig.3 Three predicted structures of one-dimensional sp3 C-H nanothreads[72]
目前,苯纳米线在实验方面的合成与分离还存在很大挑战,因此,关于纳米线性能的研究主要集中在理论计算方面。在力学性能方面,Roman 等[73]通过分子动力学计算确定苯纳米线的硬度为850 GPa,强度为26.4 nN,相对拉伸长度达14.9%,抗弯刚度为5.35×10-28N·m2。纳米线的一维结构使其韧性超过了纳米管和石墨烯,达到4.1×107N·m/kg。在纳米线的诸多构型中,tube (3,0)结构具有更高的强度和硬度[74]。在电学性能方面,金刚石基纳米线表现出宽带隙(大于4.0 eV)和绝缘性,且带隙的大小与结构密切相关,如tube (3,0)的带隙为3.93 eV[75], 而polymer-Ⅰ和polytwistane 具有更高的带隙(4.79 eV)[71]。在纳米线中引入不饱和键、杂原子或不同的取代基,可以有效地调控纳米线的带隙,如部分饱和的纳米线的带隙较窄,为1.8~4.0 eV[76];引入氮原子的吡啶纳米线的带隙可降至2.4~2.9 eV[77]。
吡啶纳米线性能的计算结果已经得到实验验证。2018 年,Li 等在苯研究的基础上,将吡啶纳米线缓慢压缩至23 GPa 时,发现吡啶经过一系列相变[78–79]后,在18 GPa 生成伪六方堆积的一维sp3C5NH5氮化碳纳米线[80]。同步辐射X 射线衍射和理论模拟表明,C5NH5氮化碳纳米线的结构更接近tube (3,0)(图4)。荧光实验结果显示,与碳纳米线相比,氮掺杂的金刚石基纳米线具有更长的发射波长,证明氮原子的引入可以有效减小金刚石基纳米线的带隙。总之,合成具有不同官能团修饰、原子级有序的金刚石基纳米线,进而调控纳米线的机械和电学性能,是该领域的重要发展方向之一。
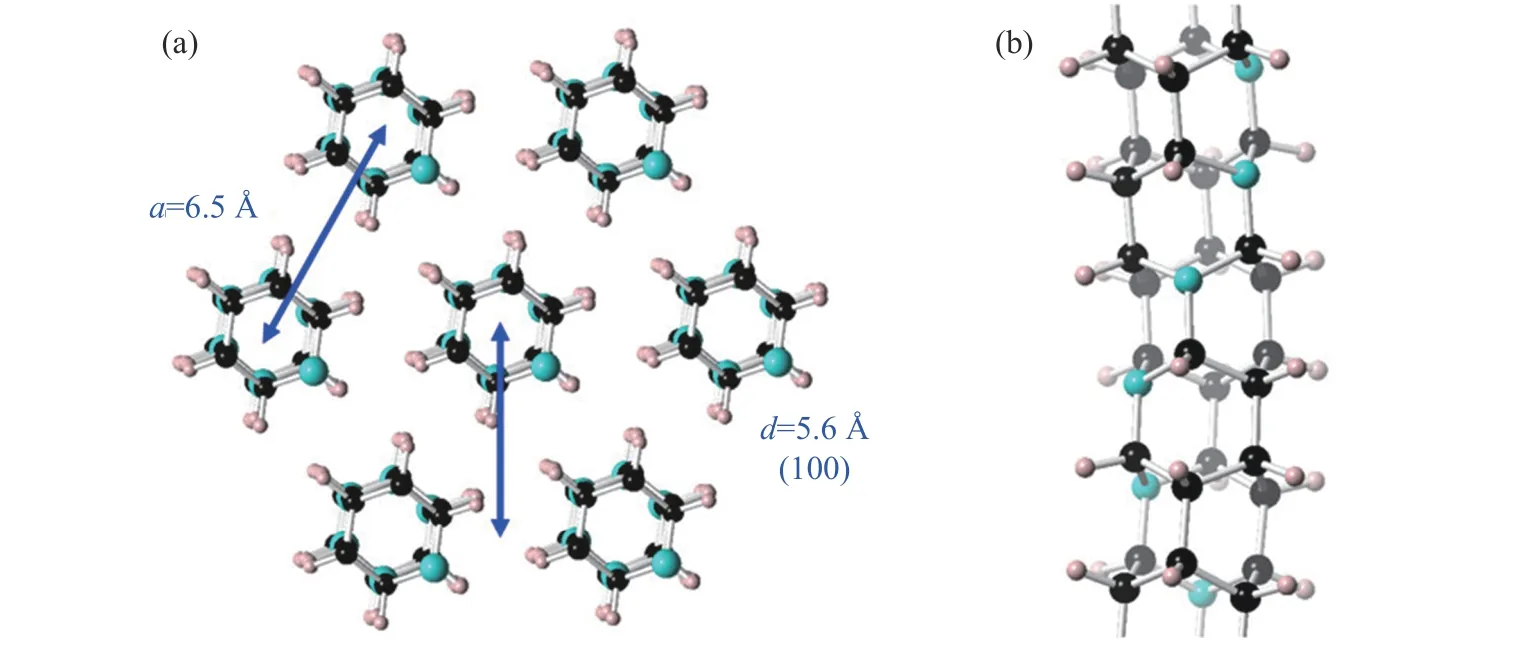
图4 沿c 轴(a)和垂直于c 轴(b)的tube (3,0)氮化碳纳米线的结构[80]Fig.4 Structure of tube (3,0) carbon nitride nanothreads viewed along the c-axis (a)and perpendicular to the c-axis (b)[80]
实现具有特定结构的原子级有序金刚石纳米线合成是实现进一步应用的重要前提。其中一个重要策略是通过引入反应活性较弱的杂原子来减少芳香分子中可能的反应路径,提高反应的选择性,进而提高金刚石基纳米线的有序性。之前的研究表明,芳香小分子在高压下聚合形成纳米线的主要反应是[4+2]环加成反应[67–69]。五元杂环芳香化合物只存在一种[4+2]加成路径,因此,可能成为生成有序金刚石纳米线的有效前体。作为一种常见的五元杂环芳香化合物,噻吩的原位单晶X 射线衍射以及振动光谱结果表明:噻吩在0.4 GPa 时从液相转变为正交固相Ⅲ,空间群为Pnma[81–82];在7 GPa 以上转变为单斜相Ⅴ[83],颜色变为黄橙色(图5(a));在20 GPa 以上,噻吩发生聚合反应,最终从35 GPa 回收得到深棕色固体产物,产率为11%左右。X 射线衍射结果表明,深棕色固体为取向有序的一维sp3噻吩基纳米线,其堆叠方式不是伪六方堆积,而是对称性更低的单斜堆积[83]。该产物与以[4+2]环加成反应为基础构建的一维噻吩衍生纳米线的结构模型有良好的一致性(图5(b))。X 射线光电子能谱和红外光谱结果表明,产物主要是噻吩环反应形成的sp3碳结构,验证了产物为一维噻吩衍生纳米线。
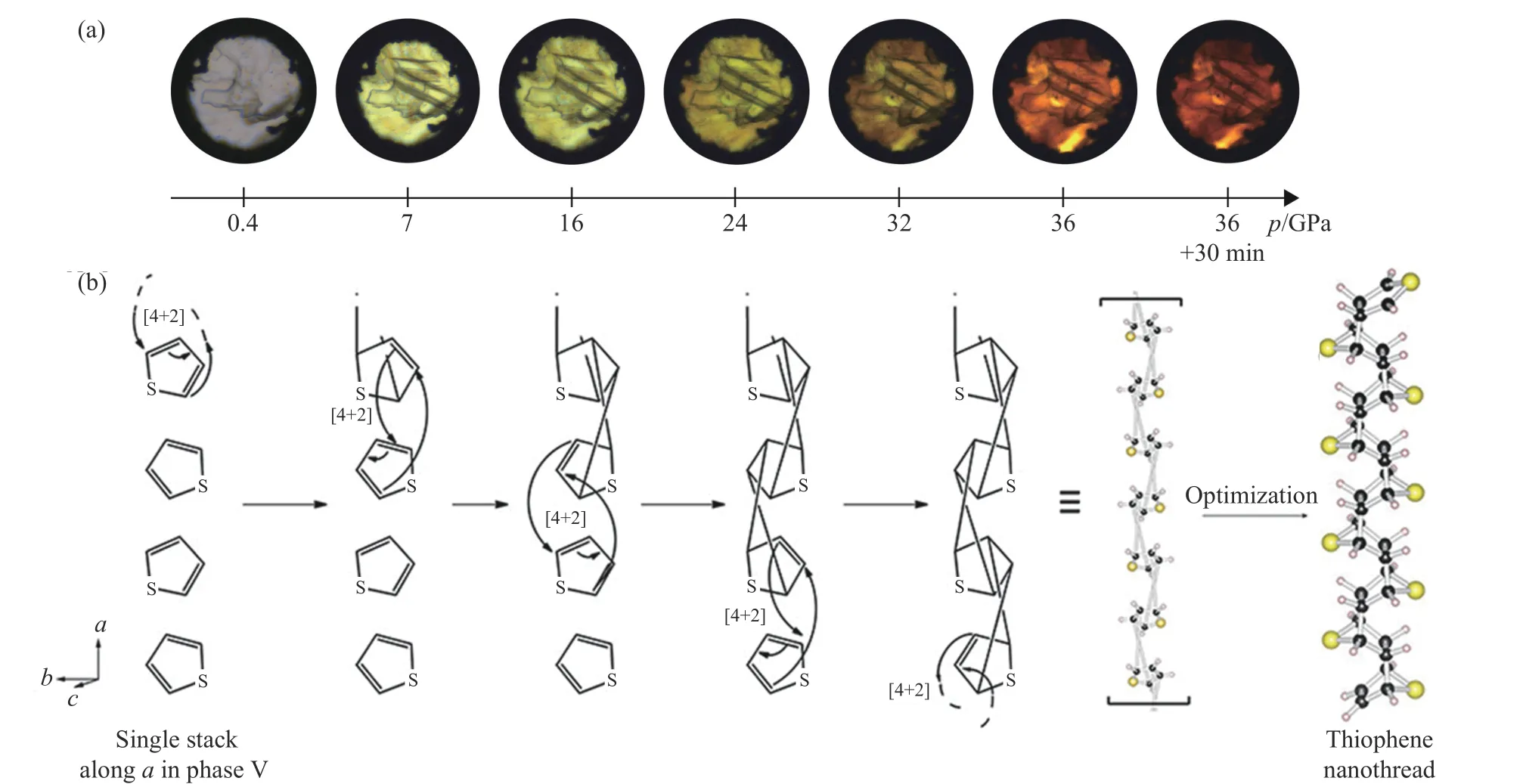
图5 (a)噻吩的光学显微照片[83];(b)噻吩通过[4+2]Diels-Аlder 反应生成反式-噻吩衍生纳米线的反应机制[83]Fig.5 (a) Optical micrograph of thiofuran[83]; (b) mechanism of formation of anti-thiophene-derived nanothreads by [4+2] Diels-Аlder reaction[83]
同样,呋喃纳米线的形成也始于呋喃分子之间的[4+2]环加成[67–69]。呋喃的芳香性较弱,因此,[4+2]加成反应的能垒较低,纳米线的合成压力也降低。之前的研究表明:呋喃在1.2 GPa 时固化,在3 GPa 左右发生相变,在10 GPa 以上反应生成黄褐色的非晶态物质[84];激光可以使反应压力降低至3.5 GPa[85]。Huss 等[86]将呋喃缓慢加压至15 GPa 得到有序的一维呋喃纳米线。原位粉末X 射线衍射结果表明,呋喃在10 GPa 时开始反应。在降压至1.5 GPa 的过程中,可以观察到一个清晰的伪六方衍射图样,证明呋喃纳米线具有有序性。当压力降至常压时,伪六方衍射图样消失,表明呋喃纳米线的有序性降低。通过[4+2]环加成反应模拟可以得到顺式、反式(氧原子朝向相反)和顺/反式呋喃纳米线的结构模型,在此基础上计算了3 种纳米线在不同堆积方式下的特定晶面间距,并与实验结果进行对比,发现反式纳米线堆积结构的晶面间距与实验结果吻合得最好,但衍射对比结果不能排除纳米线中顺/反式结构的存在。另外,计算出的3 种纳米线的红外振动模均可在实验光谱中找到。总之,实验与计算结果表明,生成的呋喃纳米线可能是反式和顺/反式等不同类型纳米线结构的混合物。之后,同位素富集的呋喃纳米线的固体核磁共振结果确定了产物主要为反式纳米线,但高度有序、完全饱和的纳米线片段大约只占10%[87]。
对于六元杂环芳香化合物,Dunning 等[88]用两个活性较低的N 原子取代苯环中相邻的两个碳原子来限制环加成反应的路线,预测并合成了哒嗪(C4H4N2)衍生的碳纳米线。哒嗪呈柱状堆积,适合作为纳米线的反应前体。理论计算表明,在30 GPa、500 K 下,哒嗪晶体相邻环上的碳通过[4+2]环加成发生自发反应,生成由sp3碳主链和顺式氮氮双键(N=N)基团组成的纳米线(图6)。原位振动光谱和X 射线衍射结果表明,哒嗪在0.2 GPa 时固化,在13 GPa 以上开始发生不可逆的聚合反应生成纳米线。回收产物的X 射线衍射结果显示,聚哒嗪产物在晶面间距d=1.2 Å处存在衍射峰,表明哒嗪纳米线为原子级有序。通过密度泛函理论计算构建了产物的顺式氮氮双键(N=N)纳米线模型,模型的计算结果与红外和X 射线衍射等实验结果吻合得较好。另外,将聚哒嗪在250 ℃下加热72 h 后发现,偶氮基以N2形式被除去,这一结果也验证了产物模型。
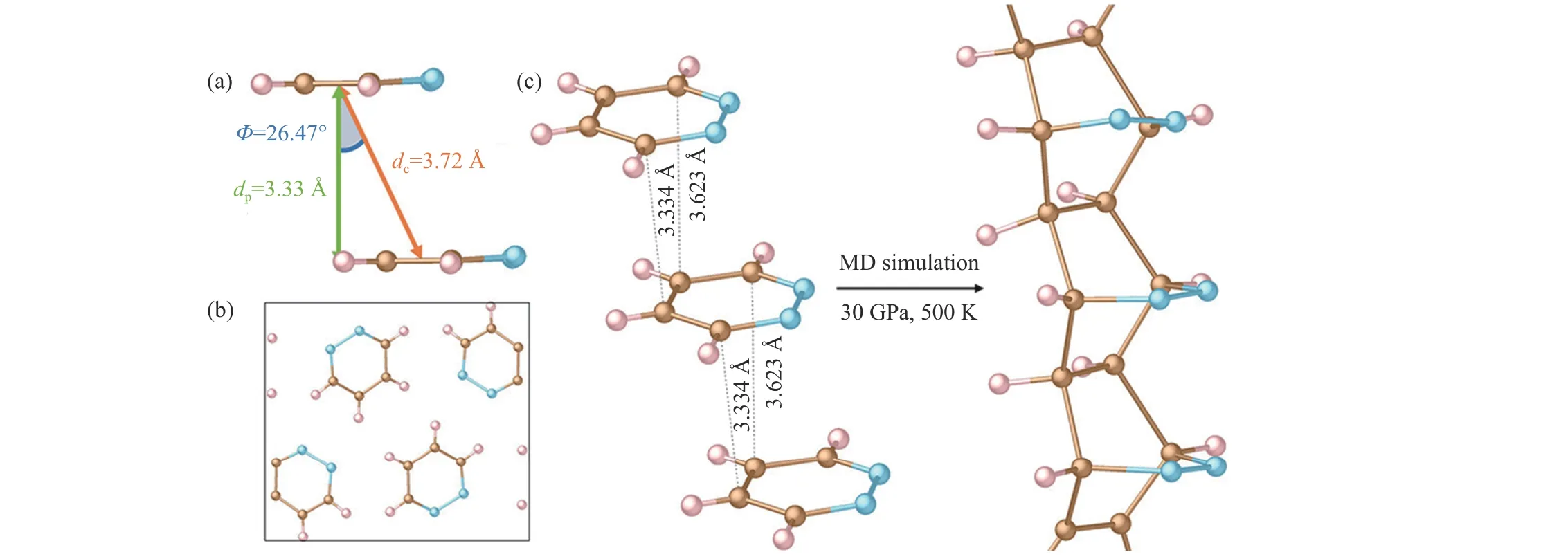
图6 (a) 哒嗪在0.61 GPa 时的局部π 堆积结构[88](dc 为哒嗪环的质心距离;dp 为平行平面之间的距离;Φ 为哒嗪环之间的滑移角,由环的法向量和质心向量确定);(b) 哒嗪沿a 轴的堆积[88];(c) 哒嗪在0 GPa、500 K 下的反应示意图[88]Fig.6 (a) π-stacking structure of pyridazine at 0.61 GPa[88], in which dc is the centroid distance between pyridazine rings, dp is the distance between parallel planes, and Φ is the slippage angle between pyridazine rings,defined by the ring normal and centroid vectors; (b) the stacking of pyridazine along a-axis[88];(c) diagram of the reaction of pyridazine at 30 GPa and 500 K[88]
几乎在同一时间,s-三嗪(C3N3H3)作为反应分子合成出了高度有序的纳米线[89]。s-三嗪在垂直于分子平面的方向上具有完美的π···π 堆积,并且分子上的N 原子与相邻分子上的C 原子正对,有利于提高反应的选择性[90]。原位X 射线衍射以及振动光谱结果表明,室温下s-三嗪在11 GPa 以上发生反应,生成有序性较差的碳纳米线,仅在晶面间距d=4.9 Å处观察到衍射峰(图7(a))。在10.2 GPa 时,将s-三嗪加热到573 K,产物的结晶度明显提高,在ab平面上具有非常好的六方对称性(图7(b)),且最小晶面间距为1.8 Å。这与大多数研究只观察到纳米线之间的晶面衍射(d=5.6 Å)形成了鲜明对比,证明了该产物结构的高度有序性。进一步通过Rietveld 精修,确定了反应产物具有一致的取向,即纳米线只采取“边对边”堆积,而非“顶点对顶点”堆积。此外,在衍射图中可以观察到002 峰,意味着产物沿c轴(即纳米线轴向)也有一定的有序性。分析临界压力(12.1 GPa)下的晶体结构和质谱数据并结合理论计算发现,s-三嗪通过相邻分子中正对的C 和N 原子成键(碳氮原子间距为2.91 Å),经过沿堆积方向的[1,3,5]协同加成反应形成tube (3,0)结构纳米线。该反应过程明显有别于其他芳香体系中的[4+2]聚合反应,是高压下新的化学反应类型,被命名为“周笼反应”。总之,s-三嗪在10.2 GPa、573 K 下形成具有完美六方取向和线内长程有序的tube (3,0)碳纳米线,证明了分子的反应选择性以及平行正对堆积对有序的金刚石基纳米线的高压合成具有非常重要的意义。
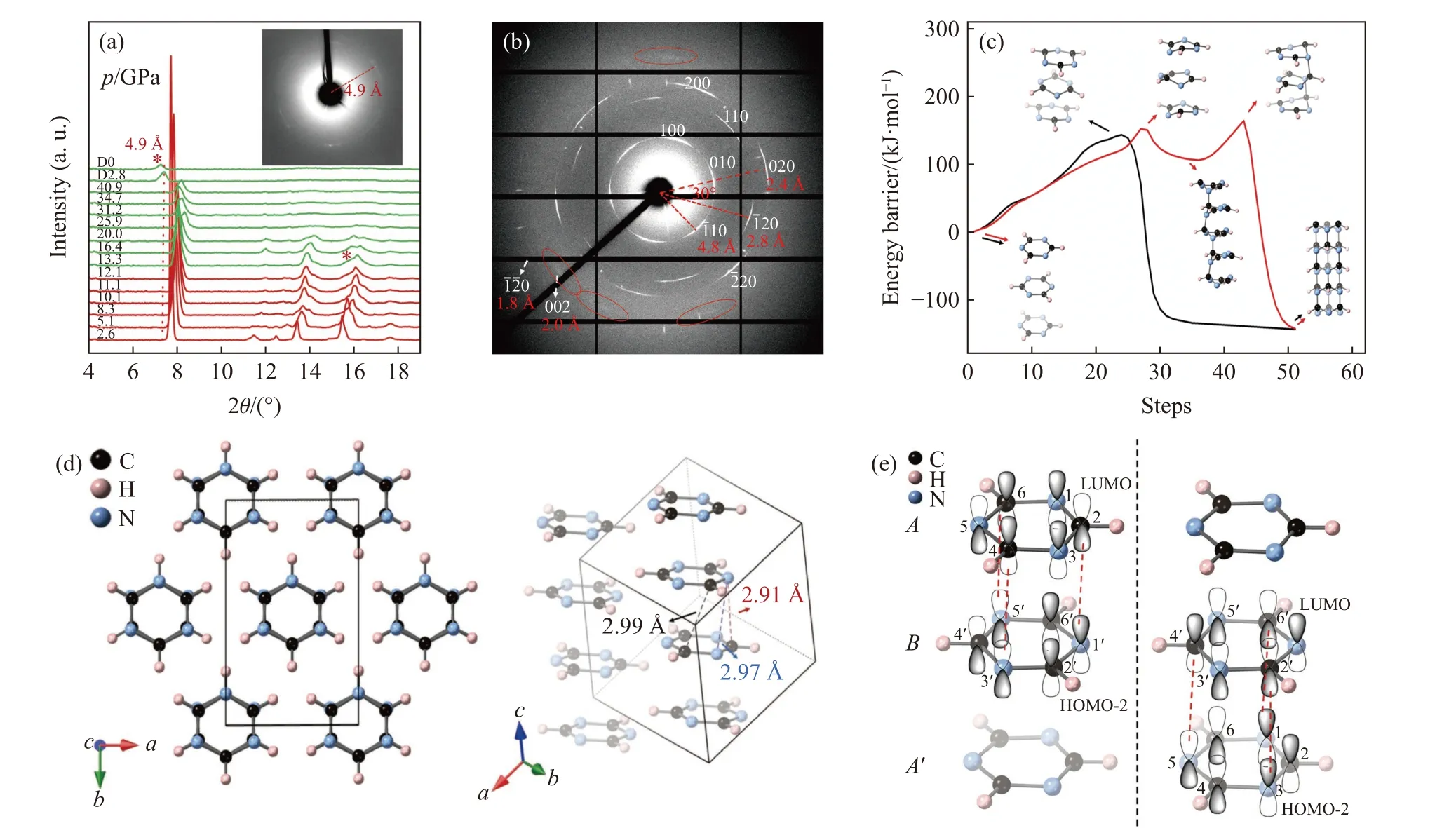
图7 (a) s-三嗪在室温高压下的原位X 射线衍射[89](D 表示降压过程);(b) 在573 K、10.2 GPa 下回收产物的X 射线衍射图像[89];(c) s-三嗪在12.1 GPa 下的晶体结构[89];(d)反应路径中各阶段的焓变(红线和黑线分别表示分步和协同反应过程)[89];(e) s-三嗪的前线分子轨道[89]Fig.7 (a) In situ XRD of s-triazine at high pressure and room temperature[89], in which D represents the decompression process;(b) XRD of the product recovered from 573 K and 10.2 GPa[89]; (c) the crystal structure of s-triazine at 12.1 GPa[89];(d) enthalpy versus step curves of each stage in the reaction path (Red and black lines represent the stepwise and concerted process, respectively)[89]; (e) molecular orbitals of s-triazine[89]
以上是通过引入杂原子限制成键路径来提高产物的有序性。除此之外,通过引入取代基、制备共晶等方法增强氢键、π···π 相互作用等分子间作用力来提高分子堆积的有序性,也是提高纳米线有序性的重要策略。
苯胺具有较强的氢键,有助于分子间的有效堆积以及分子沿堆积方向的反应。室温下,苯胺分子在被压缩至30 GPa 以上也未发生反应。这是因为苯胺分子间的氢键使相邻分子间距离变大,导致苯胺晶体相Ⅱ在较宽的温压范围内具有非常高的化学稳定性[91]。在33 GPa、550 K 下,苯胺(相Ⅱ)发生反应,生成淡棕黄色产物[92]。分子动力学研究结果表明反应生成一维产物。回收产物的红外光谱、透射电镜、X 射线衍射结果和密度泛函理论计算结果表明,苯胺沿a轴方向生成了类金刚石聚苯胺纳米线(图8(b)),产物为二维三角形堆积。值得注意的是,氢键不仅阻止了NH2基团参与反应,还有助于沿a轴方向环间C―C 键的生成,从而有助于生成有序纳米线。
同样,高的反应选择性和强氢键带来的有序分子堆积使呋喃二甲酸(2,5-furandicarboxylic acid,FDCА)成为合成高度有序碳纳米线的优异前体。通过压缩FDCА,成功合成出三维原子级有序的金刚石基纳米线[93]。原位X 射线衍射结果表明,反应在12.1 GPa 时开始,卸压至1.0 GPa 后,仍显示出一系列尖锐的衍射峰,说明产物高度有序。通过分析临界压力(10.8 GPa)下的晶体结构(图9(a))以及产物的固体核磁共振结果,可以认为FDCА 通过C2···C3′键(原子间距2.788 Å)发生[4+2]环加成反应。通过巴黎-爱丁堡压机(Paris-Edinburgh cell,PE cell)在30 GPa 下合成产物,并对其进行X 射线衍射表征,不仅发现了包括020、100 和001 峰在内的诸多尖锐衍射峰(最小晶面间距为1.4 Å),还通过Rietveld 精修直接得到了产物的原子坐标,确定其为顺式-金刚石基纳米线,直接证明了纳米线的三维原子级有序。实验及分子动力学计算表明,FDCА 的呋喃单元沿c轴发生[4+2]环加成反应生成顺式-纳米线,而氢键在ab平面上将纳米线连接成网状。此外,增加反应时间和提高温度可以显著降低FDCА 的反应压力。三维有序FDCА 纳米线的生成表明氢键对合成有序纳米线具有重要作用。
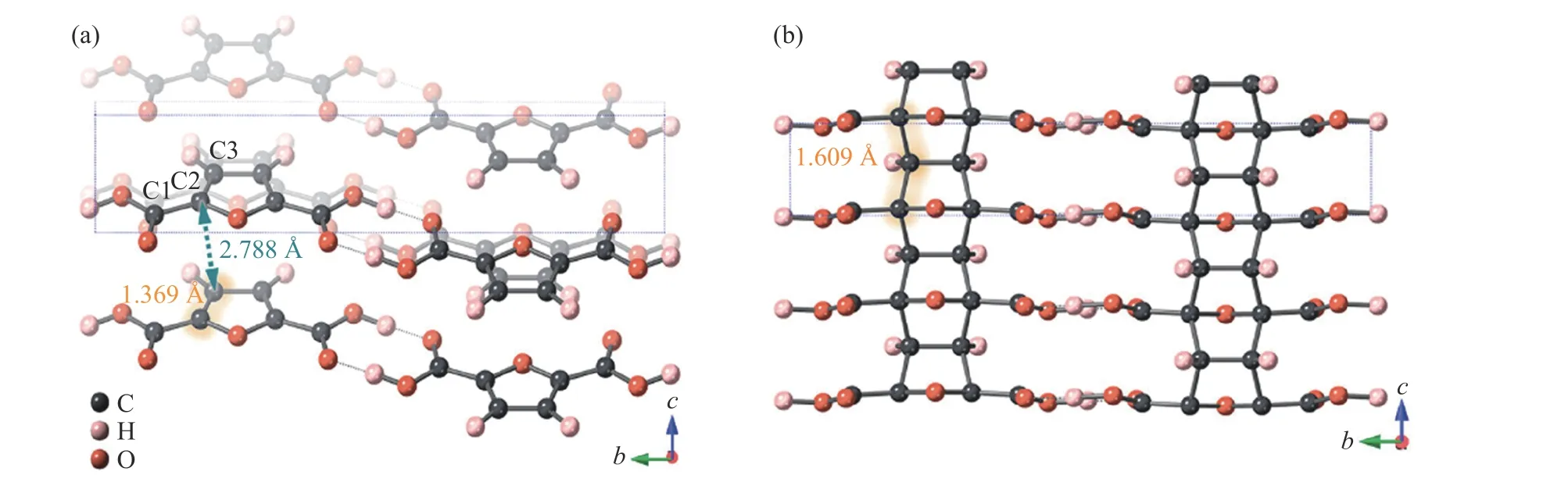
图9 (a) 10.8 GPa 时FDCА 的晶体结构[93](C1、C2、C3 为原子序号);(b) 顺式-FDCА 纳米线的晶体结构[93]Fig.9 Crystal structures of (a) FDCА at 10.8 GPa[93], in which C1, C2 and C3 are atom numbers, and (b) syn-FDCА nanothreads[93]
除了氢键之外,四极矩相反的芳烃与全氟芳烃之间会产生静电相互作用,形成非常有序的柱状堆积,限制竞争反应的发生,有利于有序碳纳米材料的高压合成。例如,在萘-八氟萘(C10H8-C10F8)共晶中,C10H8和C10F8相反的极性使这两个分子沿a轴具有近三明治状的π···π 堆积,有利于拓扑聚合的进行。研究表明,C10H8-C10F8共晶在高压下沿着分子堆叠方向(a轴)形成有序的一维sp3金刚石基纳米线[94–95]。原位单晶X 射线衍射结果显示,在20 GPa 时,C10H8-C10F8的初始衍射峰明显变宽,并出现一系列新的衍射峰,表明反应开始,进一步压缩至24 GPa 时反应完全(图10)。通过气相色谱-质谱(gas chromatography-mass spectrography, GC-MS)联用技术分析20 GPa 的回收产物发现,大部分主要峰都代表C10H8和C10F8的混合二聚体,证实了沿堆叠轴方向的聚合是主要的反应途径。为了进一步了解产物结构,Ward 等[94]根据[4+2]环加成和“2А-tube”聚合路径分别构建了产物模型(图11(b)和图11(c)),并通过密度泛函理论计算了单晶的衍射结果和红外光谱,通过与实验结果对比发现,[4+2]环加成模型与实验结果非常吻合。在该聚合路径中,沿分子堆叠方向,每个萘环上有8 个碳原子与相邻的两个分子发生4 组[4+2]反应,剩下的一个不饱和键再发生一次“拉链”反应(zipper reaction)(图11(b))。但是,Friedrich 等[95]却提出了不同的[4+2]反应路径,他们认为每个萘环上有6 个碳原子先与相邻的两个分子发生4 组[4+2] 反应,随后萘单元的两个苯环上各剩的1 个不饱和键再各发生一次“拉链”反应(图11(d))。
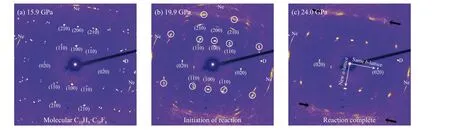
图10 C10H8-C10F8 共晶在(a) 15.9 GPa、(b)19.9 GPa 和(c) 24.0 GPa 时的单晶X 射线衍射[94]Fig.10 Single crystal XRD of C10H8-C10F8 at (a) 15.9 GPa, (b)19.9 GPa and (c) 24.0 GPa[94]
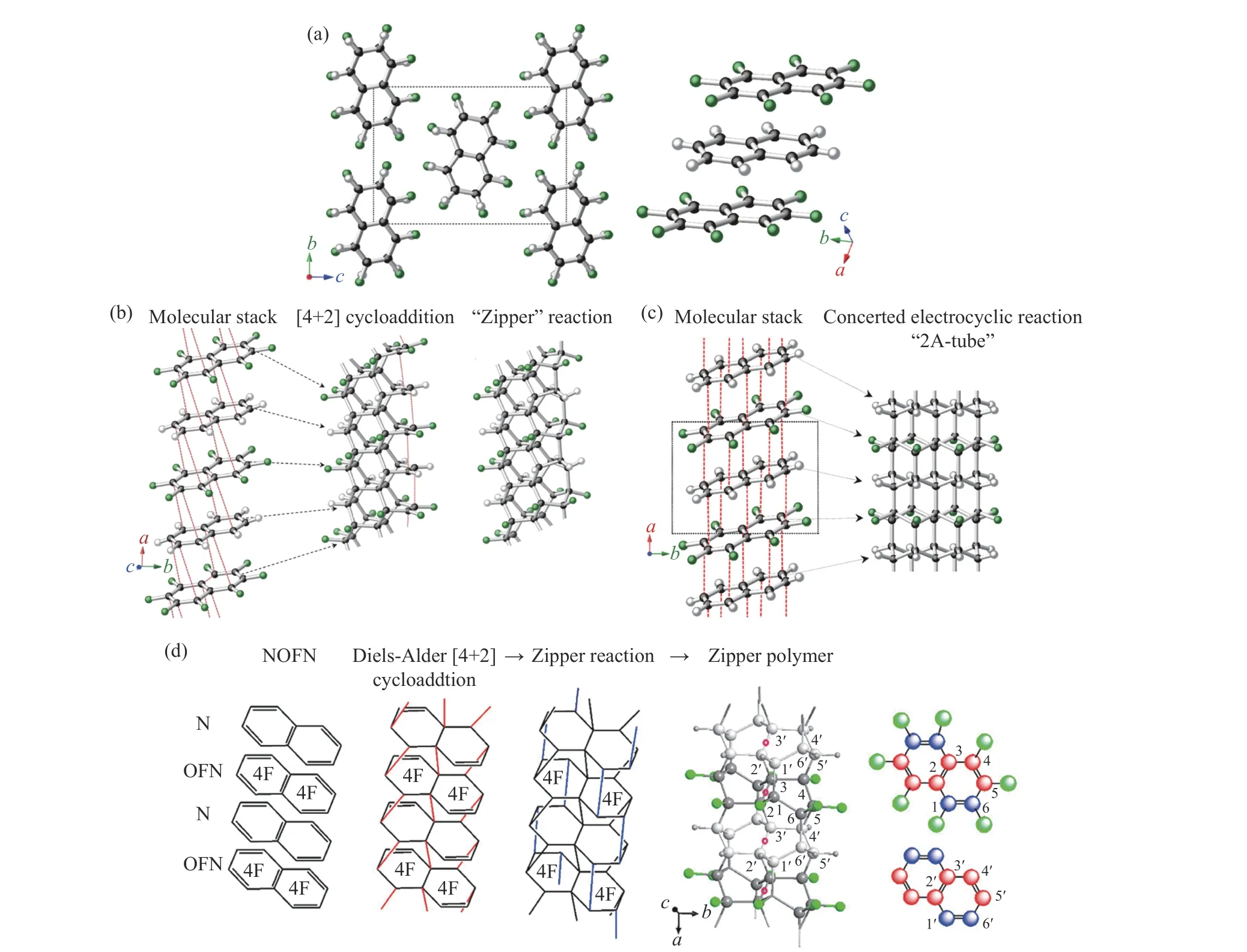
图11 (a) C10H8-C10F8 共晶的晶体结构[94];(b) [4+2]环加成聚合路径[94];(c) “2А-tube”聚合路径[94];(d) Friedrich 等[95]提出的聚合路径Fig.11 (a) Crystal structure of C10H8-C10F8[94]; (b) the [4+2] cycloaddition polymerization path[94];(c) “2А-tube” polymerization path[94]; (d) the polymerization path proposed by Friedrich et al.[95]
4 碳纳米带
在诸多碳材料中,碳纳米带有望解决二维石墨烯的零带隙问题,在电子器件等领域具有潜在的应用价值。石墨烯纳米带(graphene nanoribbon, GNR)是指宽度小于50 nm 的石墨烯条带,是一种准一维碳纳米材料。GNR 的性能与其结构密切相关,通过控制其宽度、边缘结构以及杂原子的掺杂等,可以精确地调节电子和磁性能。目前,GNR 的精准合成主要通过自下而上的合成策略实现,包括表面合成、溶液合成以及固相拓扑光热聚合3 种方法。合成一般分为两个步骤:(1)有机分子聚合成聚芳烯或其他GNR 前体;(2)石墨化,即通过环脱氢或环化生成GNR。具体而言,表面合成一般是二卤芳烃在金属表面的辅助下脱卤生成自由基中间体,并进行自由基加成聚合生成线性聚合物,然后,通过表面辅助高温退火脱氢得到石墨烯纳米带[5];溶液合成一般是芳烃单体通过Suzuki 偶联反应[6]、Yamamoto 偶联反应[7]、Diels-Аlder 反应[8]等合成聚芳基GNR 前驱体,然后,通过Scholl 反应进行环脱氢得到GNR。固相拓扑光热聚合是近年来发展的新方法。含丁二炔结构的有机分子首先在光的诱导下发生拓扑1,4-加成生成聚二乙炔,然后在高温下通过Hopf 周环和芳构化反应生成GNR[96–98]。虽然自下而上合成可以精确控制纳米带的结构,但也有不足之处[9]。比如,溶液合成法中溶解度和反应性导致的反应限制、制备过程中存在副反应等;表面合成法的反应条件严苛、反应类型有限、难以实现规模化制备等;固相拓扑光热聚合法虽然在一定程度上实现了大规模的制备,但双炔的拓扑1,4-加成反应需要合适的堆积以满足反应的几何要求(1 位和4′位炔碳原子之间的距离R1,4′< 4 Å,单体分子在堆积方向上的倾斜角 φ≈45°,具体如图12(c)所示),限制了单体的类型。高压可以有效地调控分子之间的堆积方式,压缩分子间距,在一定程度上突破分子之间的堆积限制,为纳米带的精准合成提供了新的思路。研究表明,具有适当分子结构和分子堆积的炔-苯体系,可以通过压力引发的DDА(dehydro-Diels-Аlder)反应生成碳纳米带,比如1,4-二苯基丁二炔[99](1,4-diphenylbutadiyne,DPB)和1,3,5-三乙炔基苯[100](1,3,5-triethynylbenzene,TEB)。
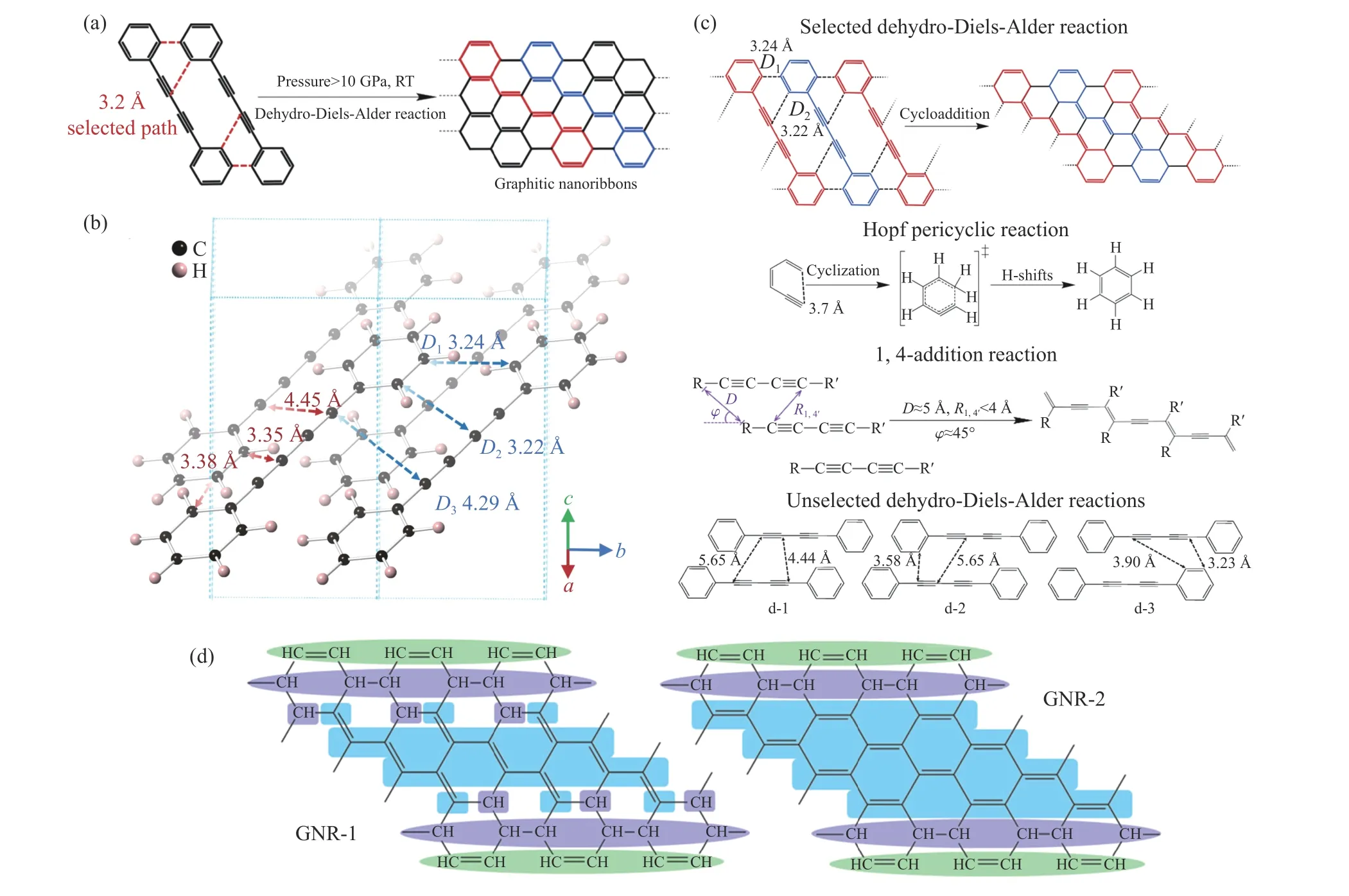
图12 (a) DPB 的反应示意图(RT 代表室温)[99];(b) DPB 在10 GPa 下的晶体结构(D1、D2、D3 为原子间距)[99];(c) 相关反应的几何结构(1,4-加成反应中,R1,4′为分子间1 位和4′位炔碳原子之间的距离,D 为取代基团(-R)之间的距离,φ为单体分子在堆积方向上的倾斜角)[99] ;(d) GNR-1 和GNR-2 的结构模型[99]Fig.12 (a) Reaction diagram of DPB, in which RT represents room temperature[99]; (b) crystal structure of DPB at 10 GPa, in which D1、D2、D3 are the distances between atoms[99]; (c) geometric structures of the relevant reactions ( In 1,4-addition reaction,R1,4′ is the intermolecular distance between 1 and 4′ alkynyl carbon atoms, D is the distance between the substituent groups (-R), and φ is the tilt angle of the monomer molecules in the stacking direction.)[99] ;(d) structural models of GNR-1 and GNR-2[99]
DPB 在10 GPa 以上发生以苯乙炔基作为双烯体、苯基为亲双烯体、距离选择性的DDА 反应,生成边缘带有sp3碳的结晶石墨烯纳米带[99](图12(a))。通过原位X 射线衍射、Rietveld 精修以及密度泛函理论计算优化,得到了DPB 在临界压力(10 GPa)下的晶体结构(图12(b))。10 GPa 时,炔基之间最短的碳碳间距为4.29 Å,大于1,4-加成的阈值间距(4.0 Å)和炔基聚合的阈值间距(3.1 Å),1,4-加成和炔基聚合反应被抑制。如图12(c)所示,DPB 最短的两个碳碳间距分别为3.24 Å (苯基上碳原子之间的距离D1)和3.22 Å (苯基碳原子与炔基碳原子之间的距离D2),相应原子排布的几何形状与Hopf 环化相似,且原子间距小于Hopf 环化发生的阈值距离(成键的活性碳原子间距为3.7 Å),因此,提出了DPB 中苯基与炔苯基之间的DDА 反应。通过分析产物的X 射线衍射、扫描电镜、透射电镜结果并结合理论模拟,可以认为,DPB 通过沿b轴的DDА 反应生成两种碳纳米带GNR-1 和GNR-2(图12(d))。GNR-1 是DPB 通过DDА 反应直接聚合的产物,GNR-2 是消除GNR-1 带内的氢原子后得到的更高共轭的纳米带。产物的中子对分布函数、红外光谱和固体核磁共振等结果均支持GNR-1 和GNR-2 的结构模型。
无独有偶,TEB 也可以通过DDА 反应生成sp2/sp3杂化的碳纳米带(图13(a))[100]。振动光谱表明,在4 GPa 时,苯基和乙炔基同时参与反应生成新的sp2和sp3碳。3.6 GPa 时的结构分析以及分子动力学计算结果表明,TEB 主要发生DDА 反应。结合从5 GPa 回收的产物的光谱、X 射线衍射、透射电镜以及核磁共振结果,确定产物主要为3-1-H、3-2-H 和3-5-H 纳米带(图13 (d))。
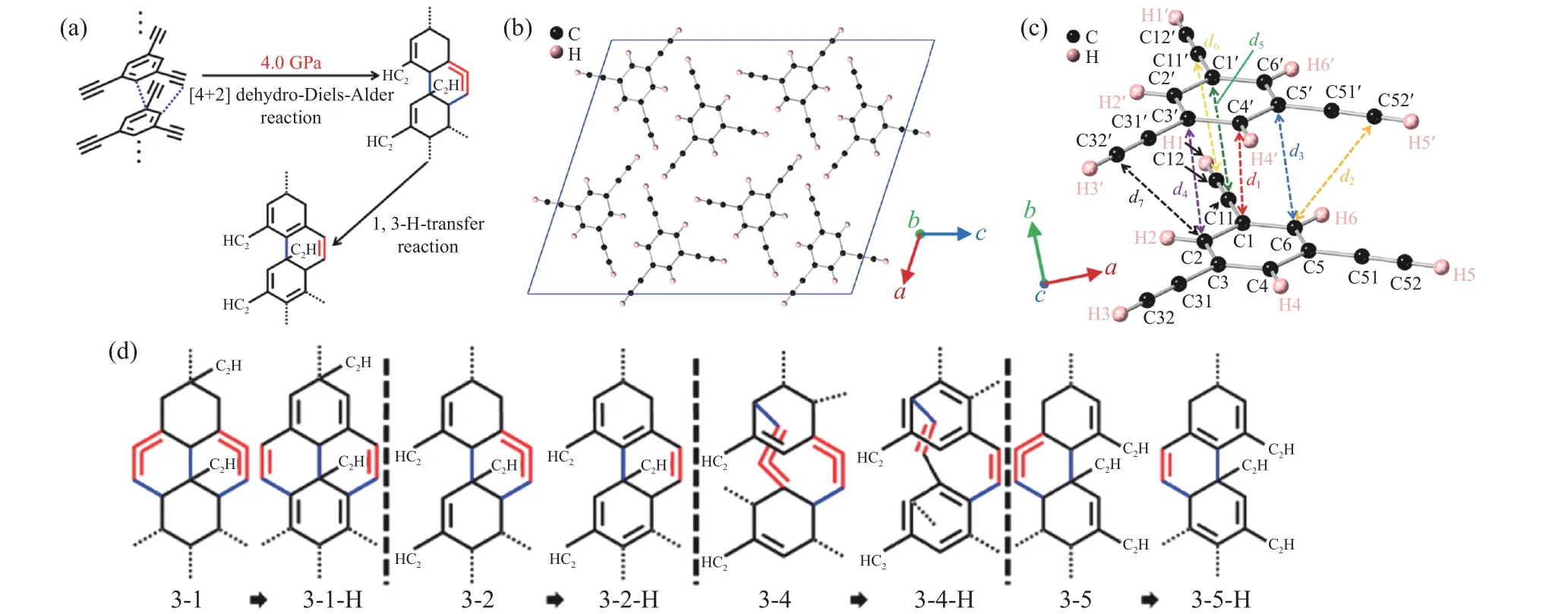
图13 (a) TEB 的反应示意图[100];(b) 3.6 GPa 时TEB 的晶体结构[100];(c) 3.6 GPa 时分子间C···C 距离[100];(d) 纳米带模型[100]Fig.13 (a) Reaction diagram of TEB[100]; (b) crystal structure of TEB at 3.6 GPa[100];(c) intermolecular C···C distance at 3.6 GPa[100]; (d) nanoribbon models[100]
除了DPB 和TEB,苯乙炔和1,4-二乙炔基苯的分子堆积不能满足DDА 反应的“距离选择”要求。因此,苯乙炔和1,4-二乙炔基苯的聚合产物都并非碳纳米带。苯乙炔在8 GPa 以上通过炔基聚合生成聚苯乙炔[101]。1,4-二乙炔基苯在10 GPa 以上发生苯基聚合生成纳米线[102]。
最近,研究发现,偶氮苯晶体在高压下也可以发生类似的有杂原子参与的Diels-Аlder 反应,生成有序的范德华碳纳米带(carbon nanoribbon, CNR)异质结。偶氮苯晶体中同时存在两种不同的分子堆积[103]。在А 层中,相邻分子并非正对堆积,而是存在空间上的滑移;在B 层中,分子是正对堆积的(图14(a))。在之前的报道中,室温、20 GPa 下偶氮苯的苯环沿b轴成键生成伪六方二维排列的双核纳米线,其中,偶氮基未参与反应[104]。而课题组的研究发现,压力会诱导偶氮苯分别在А 层和B 层中发生不同的聚合反应,生成两种碳-氮纳米带有序堆叠的范德华异质结[105]。振动光谱表明,偶氮苯从19 GPa 开始反应,偶氮基和苯基参与反应。升压至31.8 GPa 时光谱中仍存在偶氮基的振动峰,表明只有部分偶氮基团参与了反应。卸压至常压后,产物的光谱中存在明显的D 带和G 带,表明产物具有sp2-sp3石墨骨架结构。20 GPa 回收的产物的扫描电镜和透射电镜结果表明,偶氮苯沿b方向反应,形成一维碳-氮纳米带。结合临界压力(17.1 GPa)下的结构分析和理论计算结果(图14(b))可以认为:А 层偶氮苯通过偶氮苯基作为双烯体、苯基作为亲双烯体的Hetero-Diels-Аlder(HDА)反应和部分氢的消去聚合成碳氮纳米带-А(carbon-nitrogen nanoribbon-А, CNR-А),而B 层偶氮苯通过苯基的对位聚合形成碳氮纳米带-B(carbon-nitrogen nanoribbon-B, CNR-B)(图14(c))。产物的红外光谱、X 射线衍射、核磁共振的结果均支持这一模型。为了确认CNR-А 和CNR-B 的聚合路径,对25 GPa 回收的样品中的低聚物进行高分辨气相色谱-质谱联用(high resolution gas chromatography-mass spectrography,HRGC-MS)表征(图15),检测到13 种含量大于1%的化合物。其中:稠环分子(含量为8.24%)的存在证明偶氮苯在高压下发生了HDА反应,生成了产物CNR-А;联苯分子(含量为16.49%)的存在证明在高压下发生了苯的对位聚合,生成了产物CNR-B。用物质的量之比为1∶1 的C12H10N2-C12D10N2混合物进行了平行实验,产物的质谱结果与未标记的样品的产物的质谱结果相似,但存在H 和D 共标记的分子,进一步证明了该类反应发生在分子间。总之,质谱结果完全符合提出的结构模型。
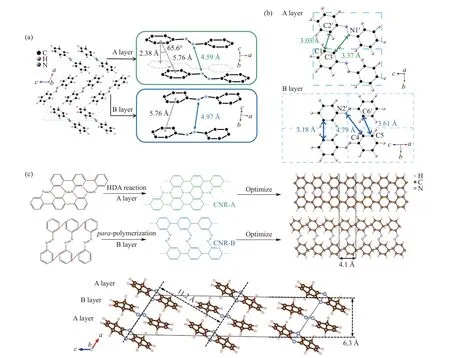
图14 (a) 常压室温下偶氮苯的晶体结构[105];(b) 17.1 GPa 时А 层和B 层中偶氮苯分子之间的距离[105](C1、C2′、C3、C4、C5、C6′、N1′、N2′为原子序号);(c) А 层偶氮苯通过HDА 反应生成CNR-А 和B 层偶氮苯通过对聚反应生成CNR-B[105];(d) 聚偶氮苯的晶体结构[105]Fig.14 (a) Crystal structure of azobenzene at atmospheric pressure and room temperature[105]; (b) the distance between azobenzene molecules in А and B layers at 17.1 GPa[105], in which C1, C2′, C3, C4, C5, C6′, N1′ and N2′ are atom numbers ;(c) the А-layer azobenzene forms CNR-А by HDА reaction and the B-layer azobenzene forms CNR-B by para-polymerization reaction[105] ; (d) crystal structure of polyazobenzene[105]
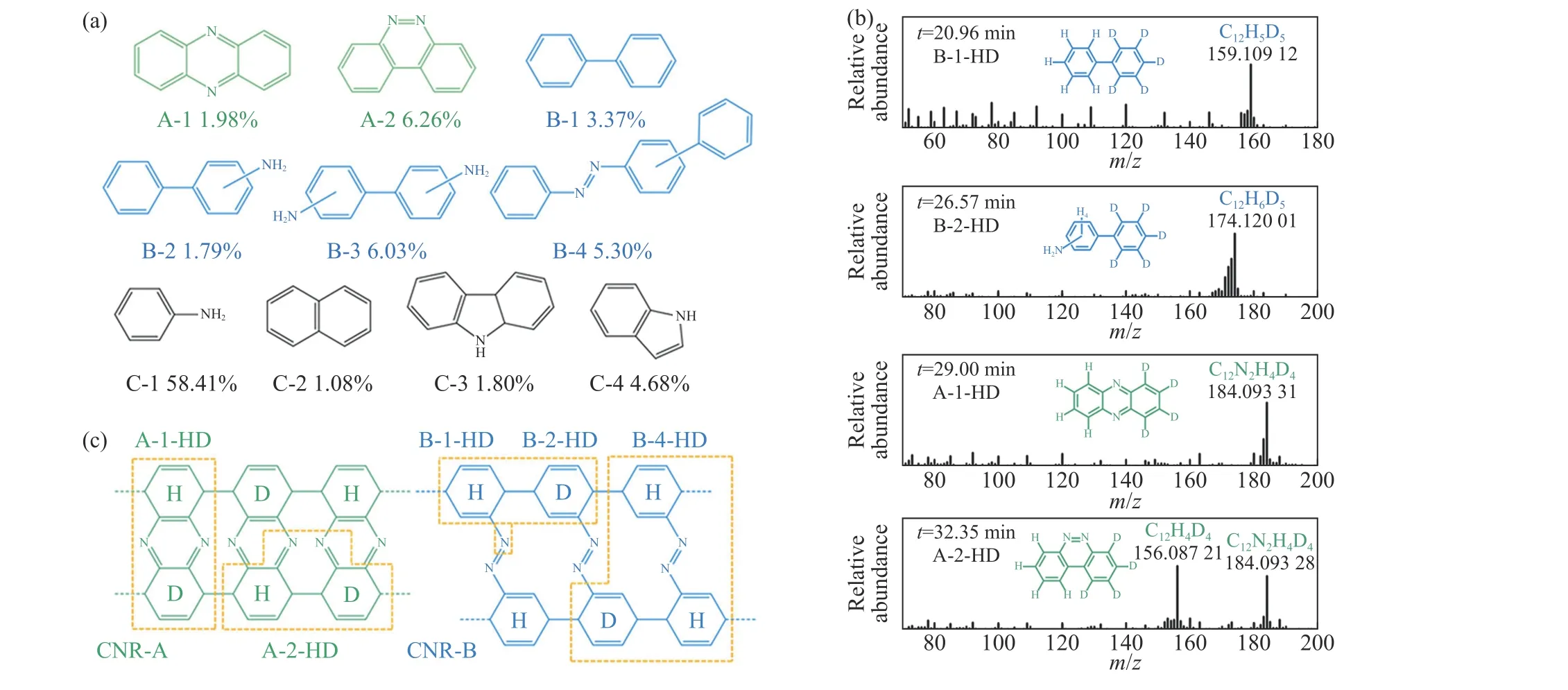
图15 (a) 通过高分辨气相色谱-质谱联用技术在25 GPa 回收的聚偶氮苯中检测到的主要低聚物及其含量[105];(b)同位素标记的低聚物的质谱和相应的分子结构(t 代表保留时间)[105];(c) 通过高分辨气相色谱-质谱联用技术在物质的量的比为1∶1 的C12H10N2-C12D10N2 混合物的聚合产物中检测到H 和D 共标记的低聚物以及这些化合物与CNR-А/CNR-B 的关系[105]Fig.15 (a) The main oligomers and their contents detected by HRGC-MS in polyazobenzene recovered from 25 GPa[105]; (b) mass spectrum and corresponding molecular structures of isotopically labeled oligomers (t represents retention time)[105]; (c) H and D colabeled oligomers detected by HRGC-MS in the polymerization products of mixture of C12H10N2-C12D10N2 with a molar ratio of 1∶1 and the relationship between these compounds and CNR-А/CNR-B[105]
5 石墨烷
在石墨烯的碳原子上添加氢原子,可将高导电的石墨烯逐渐转变为绝缘的石墨烷。通过控制反应程度可以实现对石墨烷的电子性质的调节,这使得石墨烷在储氢、压电、热电、爆炸探测和生物传感等方面具有潜在的应用前景。
乙炔是最简单的炔烃。室温下,乙炔在4.2 GPa 以上聚合生成聚乙炔[101,106]。低功率的激光可以诱导π-π*跃迁,有利于双键打开,使聚乙炔链产生支链[107]。77 K 时,乙炔在11~12 GPa、无催化剂、无光照的条件下聚合生成顺式聚乙炔,升温后转化为反式聚乙炔[108]。之前的乙炔聚合机理无法解释顺式聚乙炔的选择性。课题组通过原位中子衍射确定了反应临界压力(5.7 GPa)下乙炔的晶体结构(图16(a)),并通过分子动力学模拟成功找到生成顺式聚乙炔的反应路线,即在ac平面上,沿着a+c/a-c方向反应或者交替地沿着a+c和a-c方向反应[109]。分子动力学计算发现,顺式聚乙炔还可以进一步反应,生成短程有序的石墨烷(图16(b))。通过巴黎-爱丁堡压机分别在5 和10 GPa 下合成了乙炔的聚合产物(分别命名为P1 和P2)。红外光谱表明,P1 是以顺式聚乙炔为主的支链聚乙炔,P2 是C∶H 化学计量比为1∶1、更饱和的碳氢化合物。固态核磁共振结果表明,P2 仍然含有顺式和反式聚乙炔,并且反式聚乙炔含量相对更多(图16 (c)),表明较高的压力下,顺式聚乙炔进一步聚合生成饱和碳。同时,大量存在的sp3次甲基碳意味着发生了环加成反应,并形成了环结构。中子对分布函数结果表明,产物P2 中含有大量的碳碳单键。通过进一步与多种模型计算结果对比(图16(d)),确定了P2 为具有层状结构的环状聚合物。总之,实验结果验证了预测的反应路线:乙炔首先聚合成顺式聚乙炔(P1),然后进一步聚合成以sp3碳为主的层状多环结构(P2),类似于石墨烷。
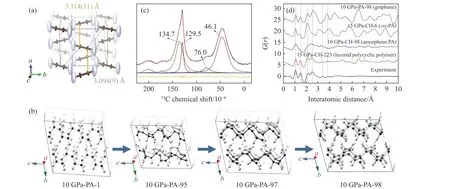
图16 (a) 5.7 GPa 下乙炔的晶体结构[109];(b) 10 GPa 时顺式聚乙炔聚合成石墨烷的分子动力学模拟(PА 代表顺式聚乙炔,PА 后的数字代表计算的代数,10 GPa-PА-1 代表10 GPa 时计算得到的顺式聚乙炔结构)[109];(c) 样品P2 的固态核磁共振[109];(d) 氘代样品P2 的中子对分布函数的实验数据和动力学模拟的结构模型的对分布函数计算结果[109]Fig.16 (a) Crystal structure of ethyne at 5.7 GPa[109]; (b) meta-dynamic simulation of the polymerization of cis-polyacetylene into graphane at 10 GPa (PА represents cis-PА, and the numbers following PА are the generation numbers.10 GPa-PА-1 is the structure of cis-polyacetylene calculated at 10 GPa.)[109]; (c) solid-state NMR of sample P2[109]; (d) the neutron pair distribution function (PDF)experiment data of deuterated sample P2 and the calculated PDF of selected structural models of the dynamic simulations[109]
不同于萘-八氟萘共晶在高压下生成一维sp3金刚石基纳米线,苯-六氟苯(C6H6-C6F6)共晶在高压下生成二维氢-氟取代的石墨烷。C6H6与C6F6之间的强静电相互作用使这两种环平行交替堆叠,有助于拓扑聚合的发生。原位光谱、X 射线衍射和中子衍射结果表明,C6H6-C6F6共晶在0.05~0.1 GPa 区间从液相转变为固相Ⅰ(R-3m),C6H6和C6F6分子交替堆叠形成柱状结构;在0.5 GPa 时,共晶从相Ⅰ转变为相Ⅴ(P21/c);1.7 GPa 时,转变为相Ⅵ;3.7 GPa 时,转变为相Ⅶ;11.3 GPa 时,转变为相Ⅷ。这些高压相与常压相不同,具有倾斜柱状结构,表明主导分子堆积的π···π 相互作用在高压下不太有效。这可能是C6H6-C6F6共晶在高压下生成石墨烷,而非一维sp3纳米线的原因之一。相Ⅷ的共晶在25 GPa 以上开始发生不可逆聚合,生成sp3(CH/F)n材料[110]。结合同步辐射X 射线衍射结果、高压下固定波长和飞行时间中子衍射结果,进一步确定了相Ⅵ、Ⅶ、Ⅷ的结构。在20 GPa 时,相Ⅷ中C6F6与C6H6分子间C···C 的最近距离约为2.8 Å,达到苯发生聚合反应的阈值距离(2.8 Å),见图17(a)。通过巴黎-爱丁堡压机在20 GPa 下合成聚合产物,并对其进行了一系列表征。扫描电子显微镜、固体核磁共振以及透射电镜结果表明,产物具有由C(sp3)、H 和F 原子构成的层状石墨骨架结构。结合红外光谱、对分布函数、选区电子衍射以及理论模拟结果,提出了短程有序的氢氟取代石墨烷的结构模型。中间产物的气相色谱-质谱联用结果表明,C6H6和C6F6在高压下的聚合反应包括[4+2] Diels-Аlder 反应、逆Diels-Аlder 反应和1-1′偶联反应,其中Diels-Аlder 反应是关键反应[111]。最终,提出了可能的反应路径,即C6H6和C6F6先通过[4+2] Diels-Аlder 反应聚合生成聚合物链,然后通过消除部分C、H、F,相邻的聚合物链进一步反应形成具有层状结构的氢氟取代石墨烷(图17(b))。
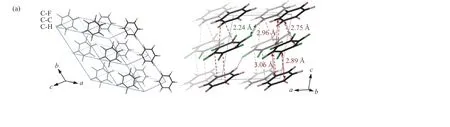
图17 (a) 20 GPa 时C6D6-C6F6 共晶的晶体结构[111];(b) 从C6D6-C6F6 共晶到H-F 取代石墨烷的反应路线[111]Fig.17 (a) Crystal structure of C6D6-C6F6 at 20 GPa[111]; (b) reaction route from C6D6-C6F6 to H-F-substituted graphane[111]
6 高电荷离子型聚合物
6.1 金属聚碳化物
压缩具有乙炔基阴离子的金属碳化物可以使原本孤立的炔基发生键合,从而合成出具有优异导电性能的金属聚碳化物材料。例如,BaC2[112]在30 GPa 以上聚合形成聚阴离子。理论研究表明,在高压下,MgC2和BeC2会聚合成碳五元环链[113];Li2C2和CaC2的炔基阴离子会聚合成一维的碳链、准二维的纳米石墨带和二维的石墨烯等结构[114–117]。课题组运用多种实验与理论计算方法,系统性地研究了CaC2在高压下的聚合反应[118],发现CaC2在18 GPa 以上发生非晶化,并伴有聚合反应的发生,电导率提高了107倍以上。进一步将高灵敏度的气-质联用手段用于高压产物的分析,测试了通过巴黎-爱丁堡压机合成的26 GPa 时的聚合产物的水解产物,发现了包括苯分子在内的数十种原料中不存在的链状和环状碳氢分子(图18(a))。这证明聚合产物中存在和其他链状或环状聚碳化物阴离子。该结果首次在实验上证明了金属碳化物中的乙炔基阴离子可以在高压下发生聚合,生成链状、环状聚合产物。30 GPa 时的分子动力学模拟结果也支持这一结论(图18(b))。此外,从水解产物中鉴定出的所有碳氢化合物的碳氢化学计量比都在1∶1 左右,证明该反应是聚合反应,而非氧化还原反应。通过进一步构建标准曲线,证明了乙炔钙在26 GPa 的聚合反应中转化率在11%以上。

图18 (a) 原料CaC2 以及通过巴黎-爱丁堡压机在26 GPa 下合成的聚合产物的水解产物总离子色谱[118];(b) 30 GPa 时分子动力学模拟的CaC2 结构[118]Fig.18 (a) Total ion chromatography of the raw material CaC2 and the hydrolysis products of polymerization product synthesized by the Paris-Edinburgh cell at 26 GPa[118]; (b) structure of CaC2 simulated by meta-dynamics at 30 GPa[118]
作为锂离子电池的电极材料,Li2C2具有非常高的理论容量,但其电导率较低。将Li2C2加压至45 GPa以上时发现电导率不可逆地提高了109倍[119]。红外光谱、拉曼光谱以及X射线衍射结果表明,Li2C2在17 GPa 时发生相变,从Immm转变为Pnma[120],在27 GPa 时开始发生聚合反应产生碳碳双键,并在35 GPa 以上进一步反应生成不可逆产物(图19(a)),与电导率的变化相吻合。为了进一步研究聚合产物,计算了相应压力范围内预测的所有Li-C 化合物[114,117,121]的红外光谱,并与实验结果进行对比(图19(b)),结果表明:在16 GPa 时,由于Li+和的重新堆积,Li2C2从Immm相转变为Pnma相;在27 GPa 以上,Li2C2开始聚合生成带状Li2C2(空间群为Cmcm),但是该结构不稳定,倾向于转化为拥有相同带结构的富碳相Li3C4。带状Li2C2在卸压过程中歧化反应加速,分解成Li6C3、Li4C3和Li3C4(图19(a))。由回收样品的水解产物的质谱分析(图19(c))可知,几乎所有的碳氢化合物都存在环状结构,证明聚合产物具有带状结构。此外,水解产物中大部分化合物的碳氢比为1∶1,但也有部分化合物的碳氢比明显偏离1∶1,意味着乙炔基阴离子发生加成聚合反应的同时也有一部分碳原子发生歧化反应。在进一步的研究中,以Lin+1C2n为基础预测了一系列不同宽度的锯齿型石墨烯纳米带(zigzag graphene nanoribbon, ZGNR),并计算了它们稳定存在的温度和压力条件(图20)[122]。在此基础上,分别在27.5 GPa、1 696 K 和36.5 GPa、2 010 K 的高温高压条件下,得到了LiC2和Li3C4两种产物。进一步的结构分析得知,LiC2具有石墨烯结构,对应于理论预测中最宽的ZGNR 结构;而Li3C4则具有聚并苯结构,是最窄的纳米石墨带。该工作为合成具有精确边缘结构和不同宽度的纳米石墨带提供了新的思路和方法。
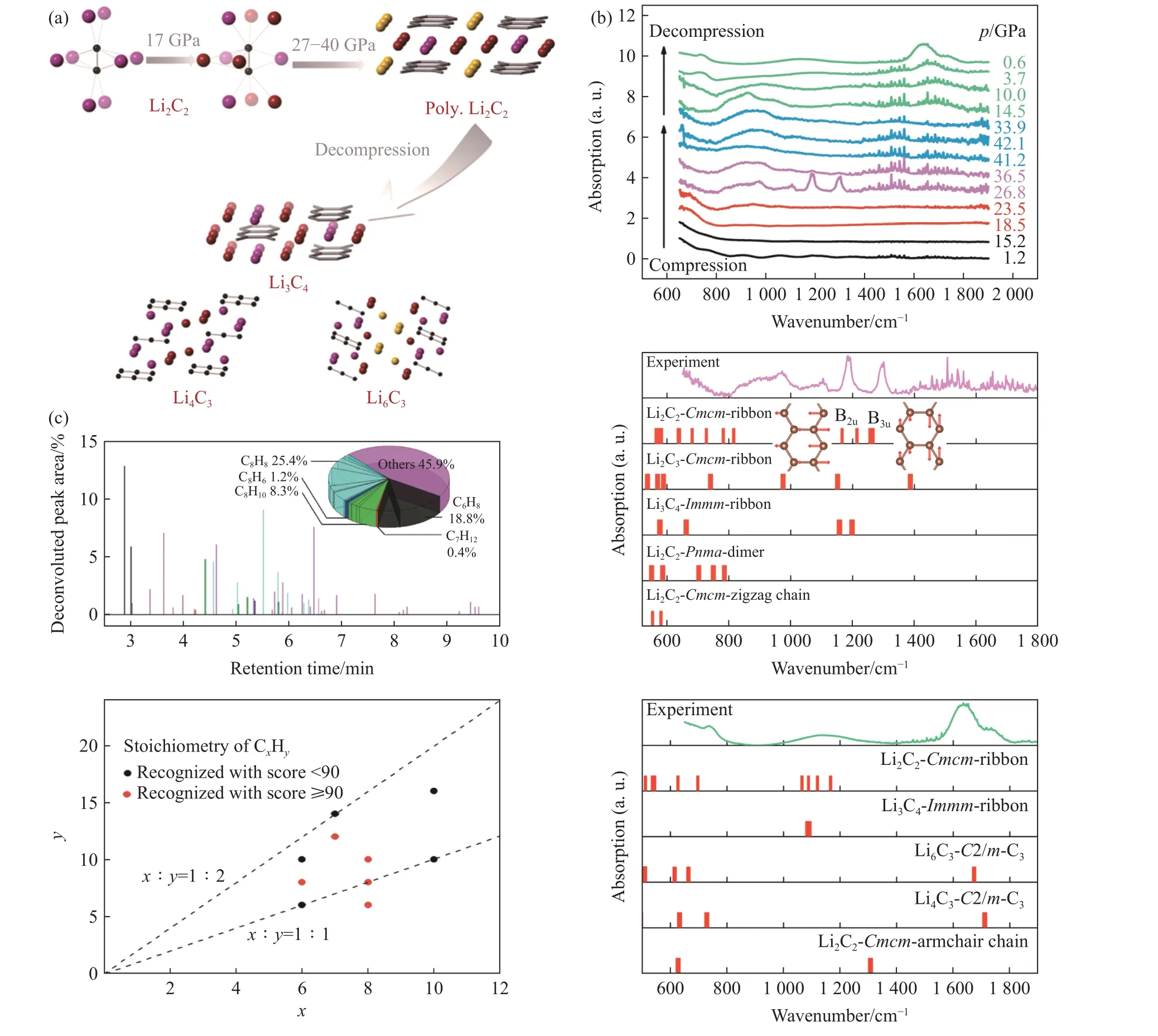
图19 (a) 高压下Li2C2 的相变及反应过程[119];(b) Li2C2 在高压下的理论和实验红外光谱[119];(c) 水解产物中各种碳氢化合物的相对含量以及化学计量比[119]Fig.19 (a) Phase transition and reaction process of Li2C2 under high pressure[119]; (b) theoretical and experimental infrared spectra of Li2C2 under high pressure[119]; (c) relative content and stoichiometry of hydrocarbons in the hydrolysis product[119]

图20 (a) 准谐波近似(quasi-harmonic approximation ,QHА)理论计算得到的Li-C 相图[122](nLi/(nLi+nC)表示Li在Li-C 化合物中的比例);Li2C2 在(b) 27.5GPa 和(c) 36.5 GPa 下的高温原位X 射线粉末衍射谱[122];(d) 27.5 GPa、1 696 K 时LiC2 和(e) 36.5 GPa、2 010 K 时Li3C4 的Rietveld 精修结果[122]Fig.20 (a) Phase diagrams of Li-C calculated by quasi-harmonic approximation (QHА)[122], in which nLi/(nLi+nC) represents the proportion of Li in Li-C compound; in situ high temperature X-ray powder diffraction pattern of Li2C2 at (b) 27.5 GPa and(c) 36.5 GPa[122]; rietveld refinement results of (d) LiC2 at 27.5 GPa, 1 696 K and (e) Li3C4 at 36.5 GPa, 2 010 K[122]
以上金属碳化物的聚合压力都比较高,但乙炔分子的聚合压力很低(3.5 GPa)。这提示可以通过减少乙炔基阴离子的电荷促进压力诱导聚合,降低聚合压力。因此,研究了单乙炔钠(NaC2H)的高压聚合反应[123]。原位振动光谱结果表明,NaC2H 在7 GPa 时发生相变,在14 GPa 左右聚合,电导率提高了2 个数量级。NaC2H 的聚合压力明显低于其他金属碳化物,这是因为NaC2H 中的C2H-只有一个负电荷,较小的压力就可以使炔基阴离子互相靠近,最终导致聚合压力降低。因此,可以认为,电荷密度越大,聚合压力越高。原位X 射线衍射得到的晶体结构以及拓扑化学分析表明,反应很可能沿d3进行(图21(b)),反应的C···C 临界距离约为2.9 Å。通过气相色谱-质谱联用手段对20 GPa 下回收样品的水解产物进行研究(图21(c)),发现水解产生的碳氢化合物的碳氢比在1∶1 左右,但略有偏差(如1-丁炔C4H6和1,3-丁二炔C4H2),证明聚合过程中存在一定的歧化反应。这种歧化反应很可能包含了得到或失去氢原子并进行自由基配对的过程,即发生了氢转移。通过搜索美国国家标准与技术研究院(National Institute of Standards and Technology, NIST)的数据库,对比分析发现,这些碳氢化合物包括链状和环状分子,证明聚合产物中也存在链状和环状的碳骨架片段。
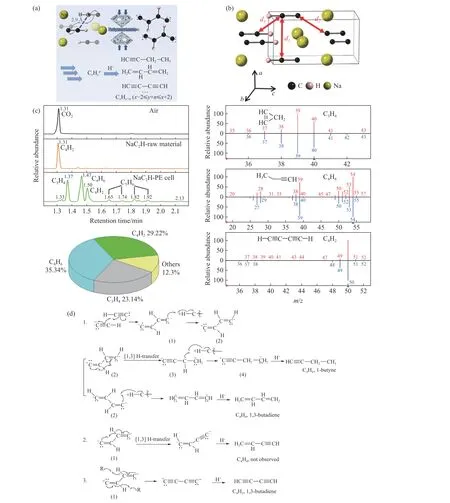
图21 (a) NaC2H 的高压反应示意图[123];(b) 临界压力下NaC2H 的晶体结构(d1、d2、d3 为原子间距[123]);(c)空气、NaC2H 以及20 GPa 下回收样品的水解产物的总离子色谱、各组分的百分比以及与NIST 谱的对比[123](m/z 代表质荷比);(d) 二聚及后续H 转移过程的反应路线[123]Fig.21 (a) Diagram of high-pressure reaction of NaC2H[123]; (b) the crystal structure of NaC2H under critical pressure[123], in which d1,d2, d3 are the distances between atoms; (c) total ion chromatography, percentages of each component and comparison with NIST spectrometry of air, raw materials and hydrolysates of samples recovered from 20 GPa[123], in which m/z represents mass-to-charge ratio; (d) reaction route of dimerization and subsequent H-transfer process[123]
6.2 聚氰化物
金属氰化物是由金属阳离子和氰基阴离子组成的离子化合物。简单金属氰化物的聚合压力很高[124–126],例如,NaCN[124]在25 GPa 以上发生聚合反应生成碳氮(C=N)双键。而过渡金属氰化物的反应压力远低于碱金属氰化物[127–130],例如,Fe[Co(CN)6][127]在10 GPa 以上非晶化。K3Fe(CN)6是配位化学和电化学中的模型化合物。课题组通过多种表征技术研究了K3Fe(CN)6在高压下的相变以及聚合反应[131]。K3Fe(CN)6在常压下的空间群为Pnca(O1 相)[132],在2~4 GPa 时转化为Pnc2(O2 相),在7~8 GPa 时转化为O3 相,但空间群仍为Pnc2,在12 GPa 时开始非晶化(图22(a))。拉曼光谱结果表明,K3Fe(CN)6的相变与氰基阴离子的聚合相关,电导率随着相变的发生而变化。4 GPa 时,氰基阴离子聚合生成连接的C=N 键,共轭双键的生成使得电导率提高103倍,达到10-4S/cm。在7~8 GPa 时电导率降低,归因于O2 相到O3 相的转变。此外,通过原位中子衍射、中子对分布函数和X 射线吸收精细结构(X-ray absorption fine structure,XАFS)测定了K3Fe(CN)6的晶体结构(图22(b))和反应前后Fe 的电子结构,结果显示Fe(Ⅲ)使CN-之间的距离更短,可以在较低的压力下成键。两个相同的 Fe(CN)36-的氰化物阴离子在碳原子之间可以形成弱的可逆键。在7~8 GPa 时,电子从CN-转移到Fe(Ⅲ),降低了氰化物离子的电荷密度,稳定了反应产物。简而言之,过渡金属可以代替碱金属,通过拉近不饱和阴离子之间的距离,降低电荷密度,使K3Fe(CN)6在较低的压力下聚合[133]。
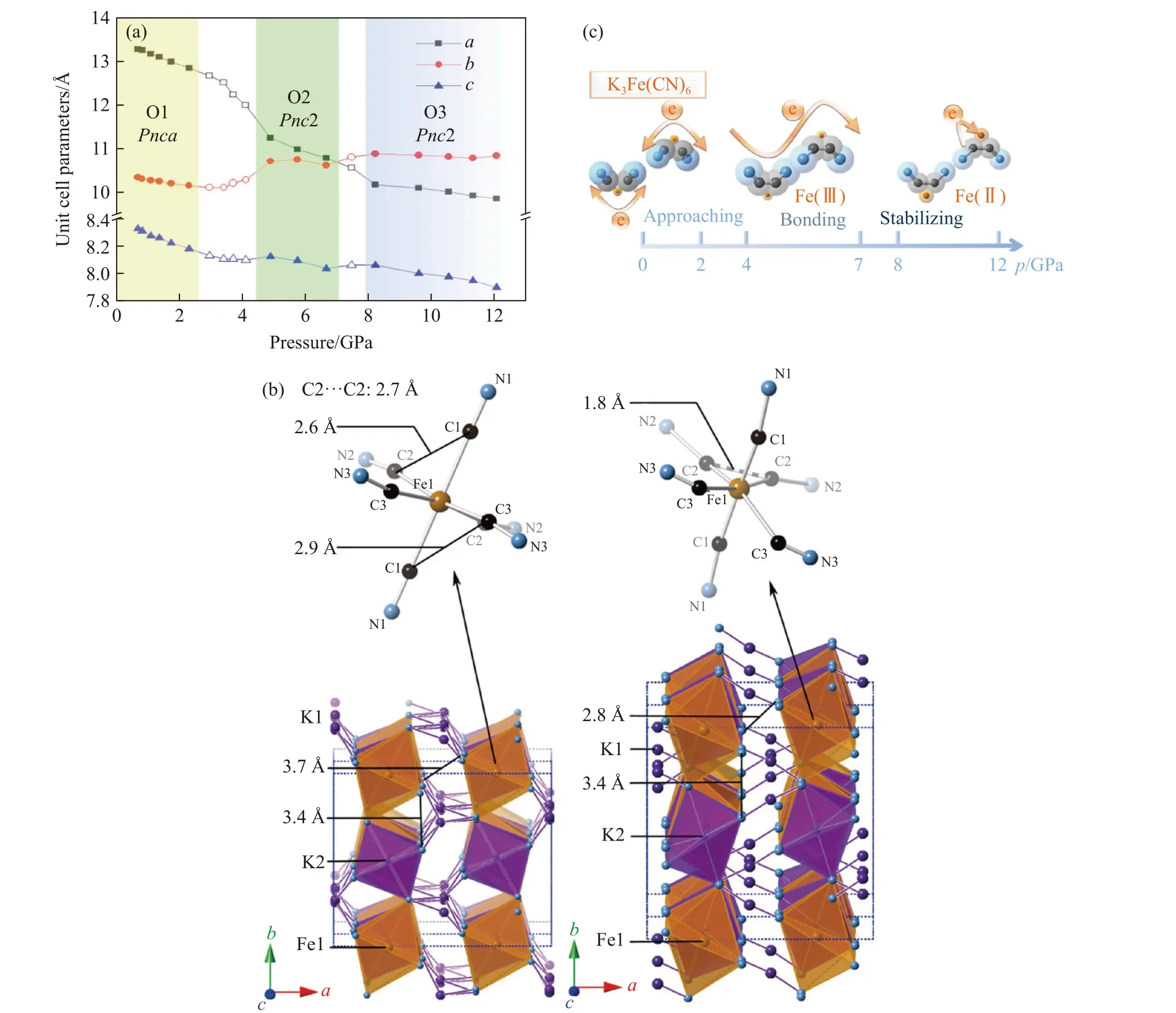
图22 (a) 高压下 K3Fe(CN)6 的晶格参数[131]; (b) 1.7 GPa(左)和4.4 GPa(右)时K3Fe(CN)6 的晶体结构和 Fe(CN)36-的局部结构[133];(c) 高压下K3Fe(CN)6 的相变和化学反应过程[133]Fig.22 (a) Lattice parameters of K3Fe(CN)6 under high pressure[131]; (b) the crystal structure of K3Fe(CN)6 and local structure of Fe(CN)36- at 1.7 GPa (left) and 4.4 GPa (right)[133]; (c) phase transition and chemical reaction process of K3Fe(CN)6 under high pressure[133]
为了进一步降低氰化物的聚合压力,将K+更换为体积更小的阳离子Li+,得到Li3Fe(CN)6。在采用原位X 射线衍射、中子衍射、中子对分布函数、拉曼光谱以及阻抗谱研究Li3Fe(CN)6的压力诱导聚合过程中发现,氰化物阴离子在1 GPa左右开始反应,在5 GPa 左右发生不可逆聚合[134]。这是所有已知金属氰化物中最低的反应压力。Li3Fe(CN)6的聚合压力小于K3Fe(CN)6的原因在于Li+的体积太小,无法像K+那样将 Fe(CN)36-层隔开,阴离子之间很容易相互接触并发生反应,从而降低了反应压力(图23)。此外,通过测量Li3Fe(CN)6的电导率发现,在10 GPa 以上,Li3Fe(CN)6完成Ⅱ-Ⅲ相变后,其电导率增大,在20 GPa 以上继续增大,说明随着压力的继续升高,一些不可逆反应还在继续进行。
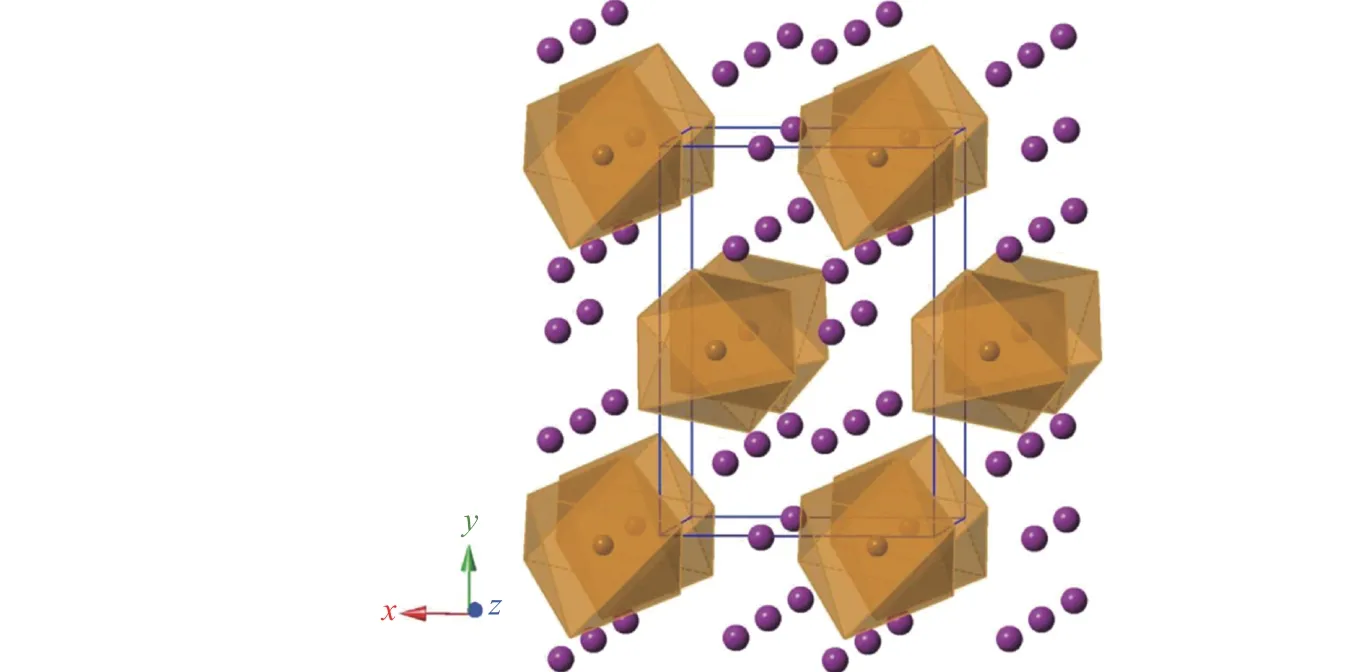
图23 Li3Fe(CN)6 的晶体结构(棕色的八面体代表 Fe(CN)36-,紫色的原子代表Li+)[134]Fig.23 Crystal structure of Li3Fe(CN)6, in which the brown octahedron stands for Fe(CN)36- and the purple atom stands for Li+[134]
7 高压反应机制
7.1 压力诱导聚合
反应物分子在高压下以固体形式存在,分子的运动受到晶格的限制。因此,高压固相聚合是一种拓扑化学反应,具有近邻优先的反应规则,产物的结构和反应路径与反应分子的堆积、分子空间距离和相对取向以及原子的反应活性密切相关。适当的分子堆积方式和较短的活性基团间距是拓扑化学反应发生的关键[135],也是高压拓扑聚合反应选择性的关键。因此,在碳基分子的高压固相拓扑聚合中,需重点关注临界压力下反应物分子的堆积和最近邻分子之间的碳碳间距。课题组总结了实验过程中以及文献报道的炔类及芳香类分子在临界反应压力时的晶体结构,发现在室温高压聚合反应中,分子间最短C···C 距离(d(C···C)min)与反应基团直接相关(见表1),即同一反应官能团具有类似的临界反应距离[136]。例如,在炔基发生聚合的高压反应中,虽然不同分子的聚合压力不同(乙炔为5.7 GPa,丁炔二酸为8 GPa),但临界压力下炔基的d(C···C)min较为一致,约为3.1 Å[61,109]。Tang 等[137]进行了乙炔晶体在高压下的分子动力学模拟、声子谱和聚合反应过渡态计算,提出“本征阈值(2.3 Å)+室温下碳原子的热振动最大值(0.8 Å)”模型,很好地解释了乙炔的经验阈值(3.1 Å)。而芳烃由于共轭结构产生的高稳定性,d(C···C)min约为2.8 Å[111,138]。Ciabini 等[138]通过对晶格声子的研究发现,苯分子高压聚合的临界反应距离约为2.6 Å,且与压力和温度条件无关。值得注意的是,部分炔基芳烃[99–100]通过DDА 反应协同成键,与炔基以及芳烃聚合的成键方式不同,所以具有较大的d(C···C)min。
7.2 氢转移反应
高压下不饱和碳基分子的聚合反应是普遍存在的。在部分腈类和炔类体系中,压力诱导聚合发生的同时还伴随着氢转移的发生,在一些反应体系中氢转移可以引发聚合反应。
氰化氢(hydrogen cyanide,HCN)是最简单的腈类分子,其特殊的分子结构使其拥有非常强的氢键。强氢键的存在使HCN 首尾相连成一条线性的链,从而堆积成四方结构[139]。对HCN 加压发现,HCN 在1 GPa 以上发生聚合反应生成黑色产物。产物为氨基和氰基修饰的C5N 稠环聚合物[140]。氨基的存在证明反应中存在氢转移过程。在乙腈分子的高压反应中也观察到了氢转移过程[141]。乙腈是典型的腈类分子。在光照条件下,氰基会发生聚合生成含有碳氮双键的聚合物。乙腈被加压至23 GPa 时生成淡黄色产物。原位红外光谱检测到碳氮双键(C=N)、碳碳双键(C=C)以及氨基,表明反应已经发生且存在氢转移过程。用巴黎-爱丁堡压机将样品加压至25 GPa,回收得到黑色固体粉末和氨气(放置在压砧周围的湿润PH 试纸在降压过程中变成蓝色;元素分析结果显示,与原料(C、N、H 的化学计量比为2∶1∶3)相比,产物(C、N、H 化学计量比为2∶0.27∶0.75)中N、H 的损失比为1∶3.09(5),该损失比与释放的氨气对应)。电子能量损失谱、拉曼光谱、对分布函数以及像差校正扫描透射电镜测量结果表明,黑色产物为洋葱状含氮纳米石墨,弯曲的层间距约为3.1 Å。固体核磁共振结果表明,该产物中含有与氮相连的碳碳双键基团(C=C(NR2)2)、亚甲基(-CH2-)、次甲基碳(H-C-R3)和与氮相连的sp3碳(NR2-CR3)等,没有氰基和甲基。这说明乙腈聚合过程中,甲基参与了氢转移反应。原位中子粉末衍射表明,在20.6 GPa 时,氘原子与氮原子(D···N)之间的最短间距为1.984 Å,比氮和氢的范德瓦尔斯半径之和小28%,意味着很有可能发生氢迁移。35 GPa 时的分子动力学计算结果表明,甲基中的C―H 键被活化,氢原子转移到氰基上,形成氨基取代的二聚体,然后进一步聚合成一维链状产物及准二维的纳米石墨带。在卸压过程中,氨基和邻近的氢原子发生消去反应,形成含氮石墨聚合物和氨气(图24)。值得注意的是,升高温度有利于这一反应的进行,从而降低反应压力,例如在250 ℃时,约7.2 GPa 的压力即可令反应发生。
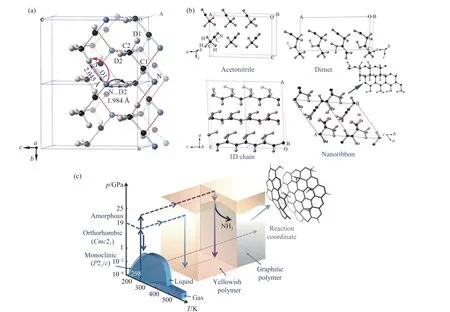
图24 (a) 20.6 GPa 时CD3CN 的晶体结构模型以及可能的氢转移路线[141](C1、C2、D1、D2 为原子序号);(b) 通过分子动力学计算得到的35 GPa 时乙腈的反应过程[141];(c) 乙腈在高压下的相变及聚合的示意图[141]Fig.24 (a) The crystal structure model of CD3CN at 20.6 GPa and possible H-transfer routes[141], in which C1, C2, D1, D2 are atom numbers; (b) the possible reaction process of acetonitrile at 35 GPa calculated by meta-dynamics[141];(c) diagram of phase transition and polymerization of acetonitrile under high pressure[141]
除了含氮体系外,氢转移反应也可以发生在纯碳体系中。研究发现,在室温高压下,固态2-丁炔(C4H6)发生分子间的C―H···π 氢转移,通过Hückel 芳香过渡态引发聚合反应生成1,3-丁二烯,而非直接进行炔基聚合(图25)[142]。原位拉曼光谱表明,2-丁炔在0.9 GPa 时固化为相Ⅰ (P42/mnm),在1.7 GPa 下转化为相Ⅱ (C2/m),在14.1 GPa 以上,相Ⅱ发生非晶化或反应,最终从18.8 GPa 回收得到无色固体产物。原位红外光谱表明,2-丁炔在11.9 GPa 以上发生构象转变,在21.2 GPa 以上sp 碳转化为sp2碳,一部分甲基转化为亚甲基(-CH2-)或次甲基(R3CH),证明发生了不可逆的氢转移和聚合。对20 GPa 下生成的无色固体产物进行质谱分析,结果表明所有产物分子的组成都是(C4H6)n,说明2-丁炔的压力诱导聚合不是传统的自由基加成聚合。通过分析低聚物的生成路径并结合光谱结果,确定1,3-丁二烯的存在,并且认为甲基参与了氢转移。在12.2 GPa 下,2-丁炔的晶体结构分析及理论计算[143]表明分子间存在非常强的CH···π 相互作用。NEB(nudged elastic band)计算发现,2-丁炔借助分子间的CH···π 相互作用,经过6 元环中间态-C2-C1-H-C2′-C1′-H′-(C2)发生双氢转移,产生1,3-丁二烯。6 元环中间态的核独立化学位移(nucleus-independent chemical shifts,NICS)结果说明,氢转移的中间态具有芳香性,且在外界压力下芳香性增强。总之,较惰性的甲基可以在高压下通过CH···π 作用发生氢转移反应,并进一步发生聚合反应。合适的分子构象和较短的H···sp-C 间距是诱导该类双氢转移反应发生的关键。
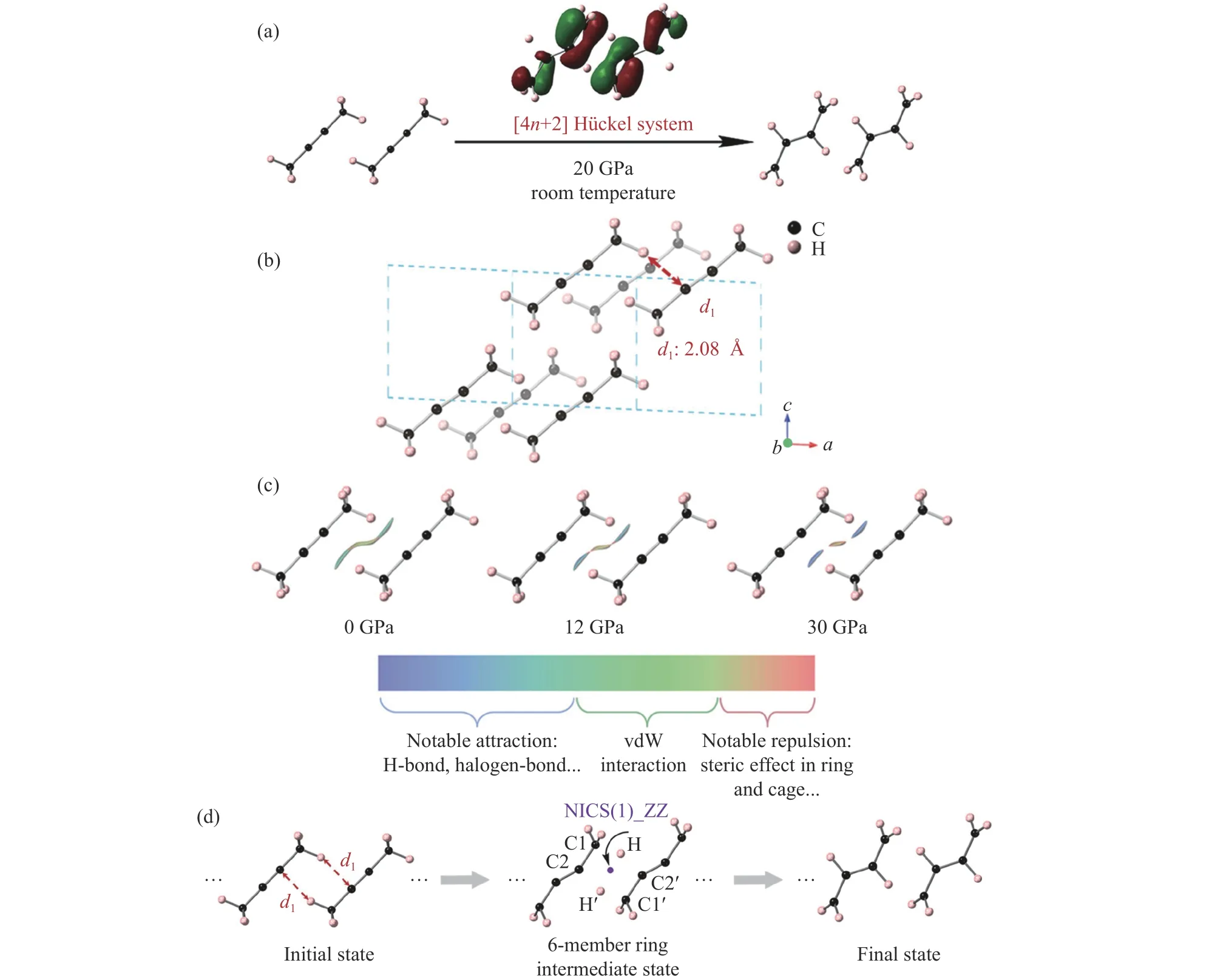
图25 (a) 2-丁炔在高压下的氢转移过程(d1 为原子间距)[142];(b)密度泛函理论计算优化后的12.2 GPa 时2-丁炔的晶体结构[142];(c) 化学键和弱相互作用的相互作用区域指示函数(interaction region indication,IRI)研究[142],IRI 为1.1;(d) NEB 计算中2-丁炔沿d1 的氢转移过程[142](C1、C1′、C2、C2′、H、H′为原子序号,NICS(1)_ZZ 表示在6 元环过渡态质心上方1 Å处环平面法向上的NICS 投影)Fig.25 (a) Hydrogen transfer diagram of 2-butylene[142], in which d1 is the distance between atoms; (b) the crystal structure of 2-butylene at 12.2 GPa after DFT optimization[142]; (c) interaction region indication (IRI) study of chemical bonds and weak interactions[142], IRI equivalent is 1.1; (d) hydrogen transfer process of 2-butylene along d1 in NEB calculation[142]( C1, C1′, C2, C2′, H and H′ are atom numbers.NICS(1)_ZZ represents NICS projection on the normal vector of the ring plane of the atom at 1 Å above mass center of the 6-member ring intermediate state.)
另外,在20 GPa、1 000 K 的高温高压条件下,由饱和碳组成的正己烷和环己烷[144]也会发生脱氢,从而生成氢化石墨。对MnOOH[145]和PdHx[146]的研究清楚地揭示了压力梯度会促使氢从高压区转移至低压区,进而驱动反应的发生。因此,认为高温对氢原子的活化作用和对样品施压时压机产生的压力梯度是促使正己烷和环己烷中的氢发生转移从而生成氢化石墨的主要原因。
8 结 论
本文主要介绍了在极端条件下通过压力诱导聚合合成的多种碳材料,包括聚烯烃、聚炔化合物、金刚石基纳米线、碳纳米带、石墨烷以及高电荷离子型聚合物。通过对碳材料合成路径的研究,发现了[4+2]环加成、[1,3,5]协同加成以及氢转移等高压反应机理,总结出临界反应距离这一高压聚合的重要参量。虽然目前大部分高压聚合还存在产物复杂、有序性不够高等问题,但高压下物质特殊的物理化学行为和高压固相反应的拓扑特性决定了高压在新型碳材料合成方面具有重要的作用和广阔的应用前景,是实现新型结构碳材料原子尺度精准合成的新兴方法。如何通过设计反应前体的分子结构和晶体堆积来控制反应路径进而合成单一有序的产物是高压合成发展的重要方向。目前,需要发展更多的原位/离位表征技术来研究高压下化合物的结构,特别是局域结构,以进一步认识高压反应机理。另外,高压下得到的具有高配位数、高电荷密度的新型碳结构材料的力学、光学、电学性质以及可能的潜在应用也是今后高压领域发展的重要方向之一。
