三联吡啶铁配位聚合物电致变色材料的合成及性能
2023-12-21陈星晗
陈星晗 束 敏 李 峰 张 蕤 刘 建*,
(1南京林业大学化学工程学院,江苏省林业生物资源高效利用协同创新中心,南京 210037)
(2潍坊科技学院,山东半岛卤水资源高值化绿色化综合利用工程技术研发中心,潍坊 262700)
0 引 言
电致变色是指材料在外部电场刺激下发生光学性质变化响应的一种现象[1]。电致变色材料通常可以在不同的氧化态或还原态之间可逆切换,从而在可见光乃至近红外区域的吸收带发生变化[2-3]。电致变色材料以其独特的光电响应特性,被广泛应用于智能玻璃、防眩目汽车后视镜、智能滤光片、电致变色显示屏、军事伪装和热控等领域[4-5]。迄今为止,电致变色材料主要包括过渡金属氧化物、过渡金属配位聚合物、紫罗精、有机共轭聚合物等[6-9]。其中,过渡金属配位聚合物电致变色材料很容易通过配体分子裁剪、金属离子选择以及配位方式变化来调控材料的电致变色性能,近年来已成为电致变色材料研究领域关注的热点之一,具有广泛应用前景[10]。
在众多的过渡金属离子中,二价铁氧化还原活性好、具有空d轨道,易与有机配体配位、成本低廉。二价铁配位聚合物被认为是一类具有广泛应用前景的电致变色材料[11]。多联吡啶铁(Ⅱ)配位聚合物通常可以由联吡啶类多齿配体与亚铁离子盐反应来制备。多联吡啶铁(Ⅱ)配位聚合物分子具有丰富的电子跃迁,在可见光区域具有很强的吸收,通常表现出紫色[12]。此外,多联吡啶铁(Ⅱ)配位聚合物在外界电压变化下,二价铁易于发生可逆氧化还原转换导致配位聚合物电子跃迁发生改变,进而产生颜色变化,是一类性能优异的电致变色材料[13]。通过在2 个三联吡啶结构单元中间引入共轭桥联基团来优化配体电子结构,这是一种调控三联吡啶铁(Ⅱ)配位聚合物电致变色材料性能的有效策略[14]。然而,目前合成的联吡啶铁(Ⅱ)配位聚合物电致变色材料通常采用有机溶剂电解液,具有一定的环境污染。因此,开发匹配水系电解质的新型联吡啶铁(Ⅱ)配位聚合物电致变色材料具有重要的学术意义和潜在的应用价值[15]。
我们通过Suzuki 偶联反应合成了3 种含有不同数量氟原子取代的苯环作为共轭桥联基团的双三联吡啶配体(F0、F2 和F4),再与四氟硼酸铁六水合物发生配位反应,制备了3 种新型三联吡啶铁(Ⅱ)配位聚合物(FeF0、FeF2 和FeF4),通过溶液旋涂法在ITO 玻璃表面成膜,并研究了其在环保型水系电解质中的电致变色性能。
1 实验部分
1.1 试剂与仪器
实验中所用到的药品、试剂及溶剂均为分析纯,所有试剂和溶剂未经处理直接使用。原料1,4-二溴苯、1,4-二溴-2,3-二氟苯、1,4-二溴四氟苯、四(三苯基膦)钯、碳酸钠、四氟硼酸铁六水合物均为市售产品。化合物1的制备参考我们之前使用的合成方法[14]。核磁表征采用美国布鲁克公司生产的Bruker DRX-600 核磁共振仪。红外光谱(FTIR)表征采用Perkin Elmer Spectrum Two 红外光谱仪。元素分析测试采用PE 2400 Ⅱ元素分析仪。X 射线光电子能谱(XPS)分析采用ThermoFisher ESCALAB250Xi型X 射线光电子能谱仪(Al 靶)。吸收光谱测试采用Perkin Elmer 公司生产的LAMBDA 365 紫外可见分光仪。电化学测试采用上海辰华公司生产的CHI-760E 电化学工作站。薄膜制备采用中国科学院KW-4A旋涂仪器。
1.2 合成步骤
1.2.1 配体F0的合成
将化合物1(1 000 mg,2.83 mmol)和1,4-二溴苯(264 mg,1.13 mmol)、碳酸钠水溶液(719 mg,2.0 mol·L-1)溶解在甲苯/乙醇(2∶1,V/V)的混合溶剂(30 mL)中,加入Pd(PPh3)4(131 mg,0.11 mmol),在氮气保护下回流过夜。反应结束后将反应混合物冷却至室温,抽滤,依次用水、甲醇、二氯甲烷洗涤滤饼,得到的配体F0 为灰色固体(555 mg,产率:71%)。化合物1 的结构经过核磁共振氢谱和碳谱表征确认。1H NMR(600 MHz,CDCl3):δ8.82(s,4H),8.76(d,J=4.2 Hz,4H),8.70(d,J=7.8 Hz,4H),8.05(d,J=7.8 Hz,4H),7.90(t,J=6.0 Hz,4H),7.83~7.81(m,8H),7.39~7.37(m,4H)。13C NMR(150 MHz,CDCl3):δ156.48,156.19,149.94,149.32,141.45,139.87,137.67,137.06,128.00,127.76,127.69,124.01,121.58,118.92。
1.2.2 配体F2的合成
将化合物1(1 000 mg,2.83 mmol)和1,4-二溴-2,3-二氟苯(305 mg,1.13 mmol)、碳酸钠水溶液(719 mg,2.0 mol·L-1)溶解在甲苯/乙醇(2∶1,V/V)的混合溶剂(30 mL)中,加入Pd(PPh3)4(131 mg,0.11 mmol),在氮气保护下回流过夜。反应结束后将反应混合物冷却至室温,抽滤,依次用水、甲醇、二氯甲烷洗涤滤饼,得到的配体F2 为灰色固体(559 mg,产率:68%)。注:该化合物溶解性很差,碳谱难以出峰,故未提供该化合物碳谱数据。配体F2 的结构经过核磁共振氢谱表征确认。1H NMR(600 MHz,CDCl3):δ8.83(s,4H),8.76(d,J=4.2 Hz,4H),8.70(d,J=7.8 Hz,4H),8.06(d,J=7.8 Hz,4H),7.91(t,J=7.2 Hz,4H),7.78(d,J=7.2 Hz,4H),7.40~7.37(m,6H)。
1.2.3 配体F4的合成
将化合物1(1 000 mg,2.83 mmol)和1,4-二溴四氟苯(346 mg,1.13 mmol)、碳酸钠水溶液(719 mg,2.0 mol·L-1)溶解在甲苯/乙醇(2∶1,V/V)的混合溶剂(30 mL)中,加入Pd(PPh3)4(131 mg,0.11 mmol),在氮气保护下回流过夜。反应结束后将反应混合物冷却至室温,抽滤,依次用水、甲醇、二氯甲烷洗涤滤饼,得到的配体F4 为灰色固体(596 mg,产率:69%)。注:该化合物溶解性很差,核磁碳谱难以出峰,故未提供该化合物碳谱数据。配体F4 的结构经过核磁共振氢谱表征确认。1H NMR(600 MHz,CDCl3):δ8.83(s,4H),8.75(d,J=4.2 Hz,4H),8.70(d,J=8.4 Hz,4H),8.08(d,J=8.4 Hz,4H),7.91(t,J=4.8 Hz,4H),7.72(d,J=8.4 Hz,4H),7.39~7.37(m,4H)。
1.2.4 配位聚合物FeF0、FeF2和FeF4的合成
等物质的量的有机配体F0 和Fe(BF4)2·6H2O 溶解在CH2Cl2/MeOH(1∶1,V/V)的混合溶剂中,在氮气保护下回流过夜。反应结束后将反应混合物冷却至室温,过滤,依次用H2O、EtOH、CH2Cl2洗涤滤饼,得到相应的聚合物FeF0,为紫色固体。以同样的方法采用F2 和F4 为有机配体分别制备配位聚合物FeF2 和FeF4。配位聚合物经元素分析表征。FeF0:(C48H32B2F8FeN6)n理论值(%):C,62.51;H,3.50;N,9.11。实测值(%):C,62.59;H,3.55;N,9.16。FeF2:(C48H30B2F10FeN6)n理论值(%):C,60.16;H,3.16;N,8.77。实测值(%):C,62.24;H,3.21;N,8.82。FeF4:(C48H28B2F12FeN6)n理论值(%):C,57.99;H,2.84;N,8.45。实测值(%):C,58.07;H,2.88;N,8.49。
1.2.5 电致变色薄膜的制备
ITO 导电玻璃依次用丙酮、乙醇、去离子水超声清洗20 min,用氮气流吹干表面的残留溶剂,放在干燥的表面皿中,继续放入臭氧清洗机中照射20 min,洁净备用。取一片洁净的ITO 导电玻璃,将导电面朝上,置于匀胶机上,用移液枪取20 μL 铁配位聚合物的N,N-二甲基甲酰胺溶液(浓度为80 mg·mL-1)于ITO 玻璃导电表面,调节转速为1 000 r·min-1,在旋涂仪上持续旋涂120 s,将所得的薄膜在加热板上80 ℃烧结10 min,烘干除去多余的溶剂,放置备用。

图示1 多联吡啶铁(Ⅱ)配位聚合物结构式和合成路线Scheme 1 Molecular structures and synthesis routes of the polypyridine Fe(Ⅱ)coordination polymers
2 结果与讨论
2.1 配体和配位聚合物的FTIR和XPS分析
对配体和配位聚合物的进行了FTIR 表征。如图1a~1c 所示,在(1 585±1) cm-1处的吸收峰归因于配体(F0、F2 和F4)中三联吡啶结构单元的C=C 键伸缩振动峰,当三联吡啶类配体与Fe2+配位形成配位聚合物(FeF0、FeF2 和FeF4)后,配位聚合物中三联吡啶中的C=C 键伸缩振动峰发生一定程度上的偏移,移动到(1 610±4) cm-1处。类似的,在(1 390±4)cm-1处的吸收峰归因于配体(F0、F2 和F4)中三联吡啶中的C—H 键伸缩振动峰,在(1 400±10)cm-1处的吸收峰归因于配位聚合物(FeF0、FeF2和FeF4)中三联吡啶中的C—H 键伸缩振动峰[16]。FTIR 特征峰的偏移充分说明Fe2+与三联吡啶配体发生了配位。此外,我们对3 种配位聚合物进行了XPS 表征。如图1d所示,位于399.8 eV 的信号峰归属于Fe(Ⅱ)配位聚合物中三吡啶基配体的N1s。配位聚合物阴离子BF4-中的B1s和F1s的信号峰分别位于193.5 和685.9 eV,配位聚合物金属中心离子Fe2+的特征信号峰Fe2p3/2和Fe2p1/2分别位于708.5和721.4 eV[17]。
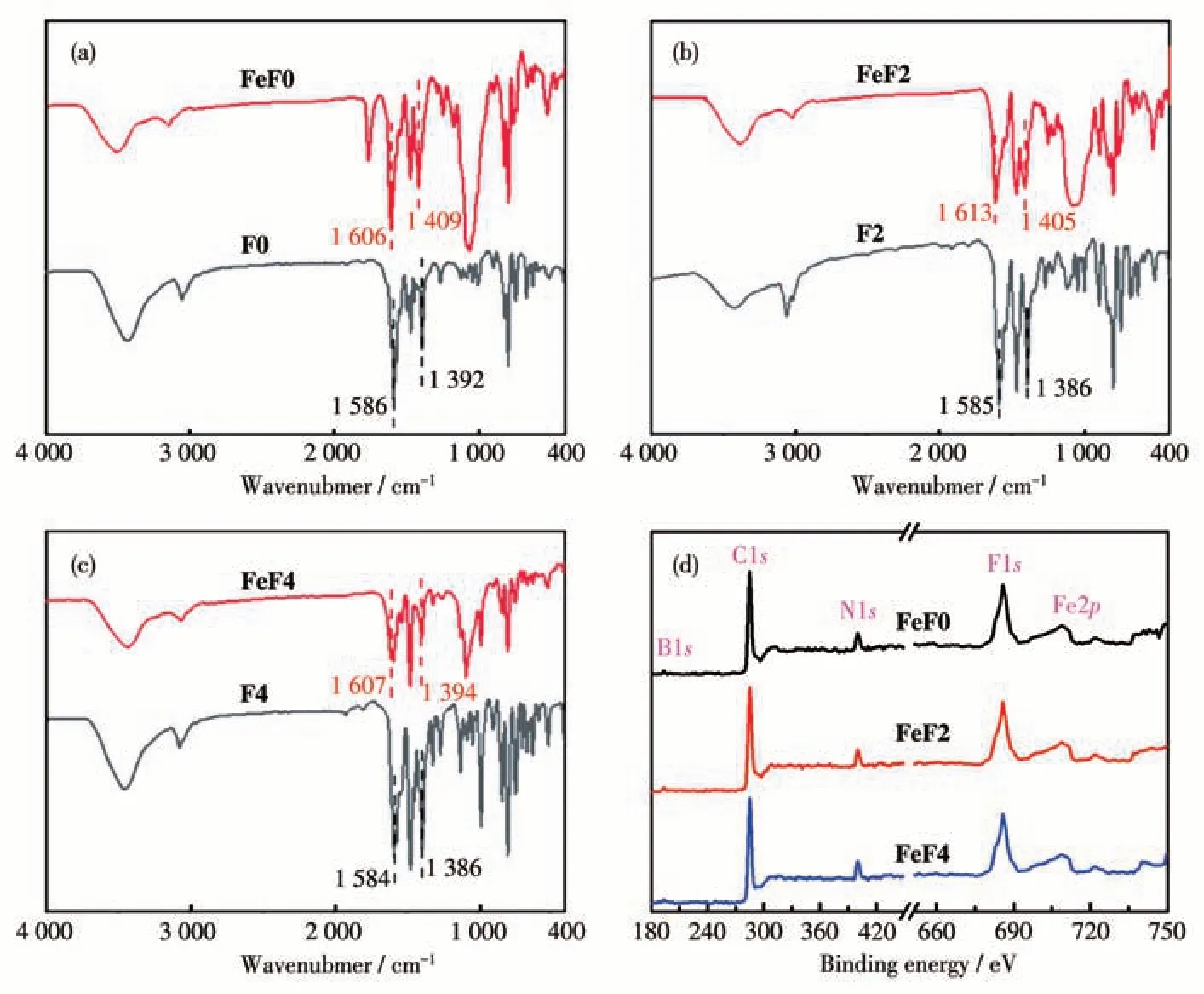
图1 三联吡啶配体和配位聚合物的FTIR谱图(a~c);配位聚合物的XPS谱图(d)Fig.1 FTIR spectra(a-c)of terpyridine-based ligands and the coordination polymers;XPS spectra(d)of the coordination polymers
2.2 配位聚合物薄膜的循环伏安(CV)测试
以表面旋涂有配位聚合物薄膜的ITO 导电玻璃作为工作电极,铂丝电极作为对电极,Ag/AgCl电极作为参比电极,0.1 mol·L-1LiBF4/H2O 作为电解液,采用三电极体系,在不同的扫描速率下(40、60、80、100、120、140 和160 mV·s-1),通过施加从0~1.2 V 的外界电压,进行CV 曲线测试,研究结构中含有不同个数氟原子取代的苯环桥联基团对Fe(Ⅱ)配位聚合物电致变色薄膜的电化学性质的影响和相关的电化学行为。如图2 所示,在不同的扫描速率下,配位聚合物FeF0、FeF2 和FeF4 在0.6~0.9 V存在一个明显的氧化还原峰,源于配位聚合物的Fe(Ⅱ)/Fe(Ⅲ)氧化还原电对。随着扫描速度的不断增加,配位聚合物薄膜的峰值电流也在不断增大,表明旋涂在ITO 表面的配位聚合物薄膜具有良好的电化学活性。图3 显示了3 种聚合物FeF0、FeF2和FeF4 在CV 曲线测试中阳极峰电位(Epa)和阴极峰电位(Epc)与扫描速率的平方根呈线性关系,且最小均方误差(R2)非常接近于1.0,这表明这3 种配位聚合物薄膜紧密均匀地附着在ITO 玻璃表面,并且通电时发生的电化学氧化还原过程不受电解液缓慢扩散的影响[18]。
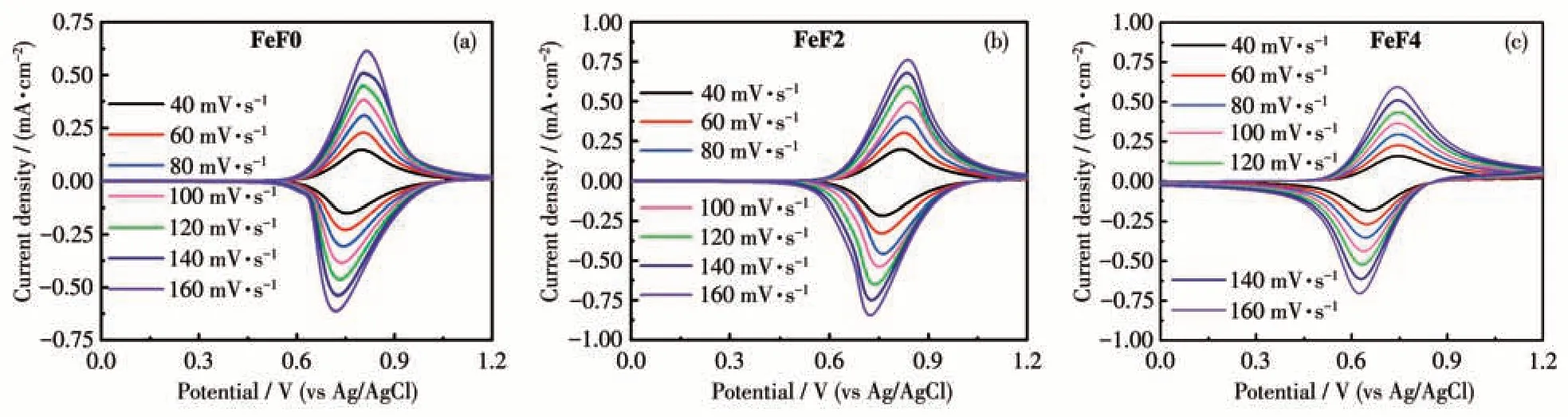
图2 40~160 mV·s-1扫描速率下配位聚合物薄膜在0.1 mol·L-1 LiBF4水溶液中的CV曲线Fig.2 CV curves of the coordination polymer thin films in aqueous solution of LiBF4(0.1 mol·L-1)at different scan rates from 40 to 160 mV·s-1

图3 对应于图2中CV曲线阳极和阴极峰值电流与扫描速率的平方根的线性关系Fig.3 Linear dependence of the anodic and cathodic peak current of the CV curves and squre root of the scan rates corresponding to Fig.2
2.3 配位聚合物薄膜的电致变色性能表征
在外加电压变化下对配位聚合物薄膜的光谱电化学性能进行了表征。以表面旋涂有Fe(Ⅱ)配位聚合物薄膜的ITO 玻璃作为工作电极,铂丝电极作为对电极,Ag/AgCl 电极作为参比电极,组成三电极体系。以LiBF4水溶液(0.1 mol·L-1)作为电解液,采用紫外可见分光光度计和电化学工作站联用,进行光谱电化学测试。图4 呈现了FeF0、FeF2 和FeF4电致变色薄膜在施加0~0.90 V、0~1.00 V、0~1.15 V电压条件下的紫外可见吸收光谱图。从图中可以看出,配位聚合物FeF0、FeF2 和FeF4 表现出相似的电致变色行为,在施加外界电压条件下,3 种配位聚合物薄膜颜色从紫色变为黄色。强吸电子取代基(氟原子)的引入会显著影响Fe(Ⅱ)配位聚合物的电化学氧化还原电位,导致氧化电位增大。随着外界电压的逐渐增大,3 种配位聚合物中典型的金属-配体电荷转移(MLCT)跃迁吸收峰575 nm 左右在0.90、1.00 和1.15 V 时明显减小甚至消失。与此同时,一个新的吸收峰出现在400 nm 附近,其吸光度随外加电压的升高而逐渐增强,导致3 种配位聚合物薄膜的颜色从紫色变为黄色。当撤掉外加电压后,3 种薄膜又变回到初始状态的紫色。

图4 不同电压下配位聚合物薄膜在0.1 mol·L-1 LiBF4水溶液中的紫外可见吸收光谱(插图为施加电压后薄膜颜色变化)Fig.4 UV-Vis absorption spectra of the coordination polymer thin films in aqueous solution of LiBF4(0.1 mol·L-1)under different applied voltages(Inset:color change of the electrochromic polymer thin films under external applied voltages)
采用双电位计时电流法对配位聚合物电致变色薄膜FeF0、FeF2 和FeF4 的光学对比度、响应时间和稳定性进行了探究。在0 V 与相应最大的氧化电压之间施加阶跃时间为5 s 的阶跃电压,记录了3种配位聚合物电致变色薄膜FeF0、FeF2 和FeF4 在波长575 nm 透过率随电压的变化曲线,并以透过率达到最大值的95%的所需时间来计算着色时间,具体数据见表1。如图5 所示,FeF0、FeF2 和FeF4 在575 nm 处光学对比度分别为50.4%、69.0% 和44.7%。此外,3 种配位聚合物电致变色材料表现出快速的响应速率。其中,FeF0、FeF2 的着色时间(tc)均为0.5 s,FeF4 的着色时间为0.4 s,略快于FeF0 和FeF2,这表明3 种Fe(Ⅱ)配位聚合物的中心离子Fe2+都能够快速氧化。FeF0和FeF2的褪色时间(tb)分别为0.9 和0.5 s,都远远短于FeF4 的褪色时间(1.9 s)。电致变色薄膜FeF0、FeF2 和FeF4 均表现出良好的循环稳定性,在经历过200 次循环后仍可保持原始最大光学对比度的90%以上。相比于FeF0,引入了2 个强吸电子基氟原子的配位聚合物FeF2 表现出更好的电致变色性能。然而,引入了4 个强吸电子基氟原子的配位聚合物FeF4 的电致变色性能相对欠佳。结果表明,聚合物引入适量的强吸电子基氟原子可以影响共轭聚合物分子内和分子间的相互作用,从而改善聚合物材料的电致变色性能。
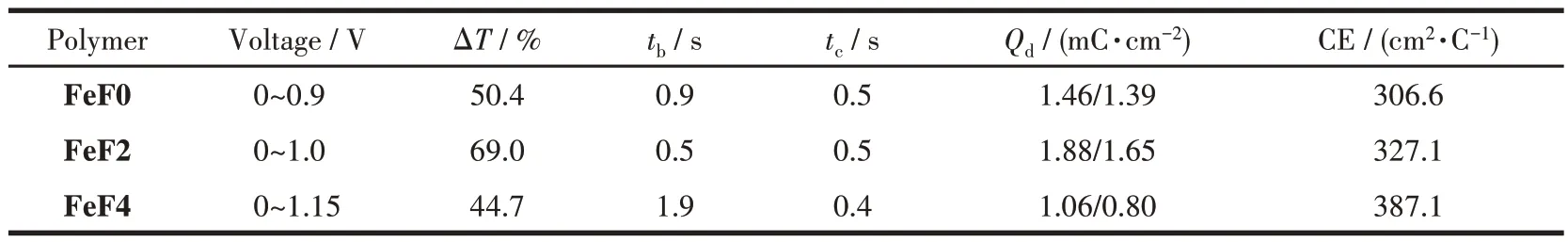
表1 配位聚合物电致变色薄膜在波长575 nm附近的电致变色性能参数Table 1 Electrochromic parameters of electrochromic thin films based on the coordination polymers monitored around 575 nm
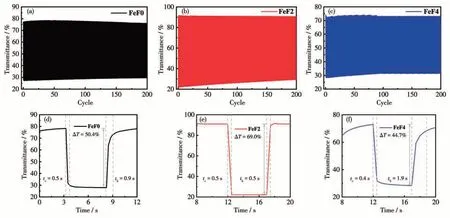
图5 配位聚合物电致变色薄膜的循环稳定性测试(a~c);配位聚合物电致变色薄膜在波长575 nm附近的光学对比度和响应时间(d~f)Fig.5 Cyclic stability test(a-c)of electrochromic thin films based on the coordination polymers;Optical contrast and response time(d-f)of electrochromic thin films based on the coordination polymers monitored around 575 nm
着色效率(CE)是评价电致变色材料能耗情况的重要参数之一,表征电致变色材料在单位电荷注入时引起的颜色变化效率。根据Lambert-Beer 定律,CE值的计算可以根据式1计算[14]:
其中,ΔOD 为光学密度的变化值,Qd为单位面积注入和抽出的电荷量,Tb为褪色态透过率,Tc为着色态透射率。采用计时电流法和紫外可见分光光度计联用对配位聚合物电致变色薄膜进行电化学表征,并通过记录电流随时间的变化来测试充电/放电量(图6)。结果表明,配位聚合物FeF0、FeF2 和FeF4 电致变色薄膜表现出较高的着色效率,分别为306.6、327.1 和387.1 cm2·C-1,具有潜在的应用价值。

图6 配位聚合物电致变色薄膜的充放电曲线Fig.6 Charge/discharge curves of electrochromic thin films based on the coordination polymers
3 结 论
通过Suzuki偶联反应以苯基或氟取代苯基作为桥联基团共轭连接2个三联吡啶结构单元,构建了3个双三联吡啶配体,进一步与四氟硼酸铁六水合物进行配位反应制备了3 种Fe(Ⅱ)配位聚合物(FeF0、FeF2 和FeF4),通过溶液旋涂法在ITO 导电玻璃表面旋涂成膜,并研究了薄膜在水系电解质体系中的电化学性质和电致变色性能。3 种配位聚合物FeF0、FeF2 和FeF4 电致变色薄膜均表现出快速的紫色-浅黄色的颜色切换速率(着色时间和褪色时间均低于1 s),较高的着色效率(均超过300 cm2·C-1),以及良好的循环稳定性。配位聚合物FeF0、FeF2和FeF4 电致变色薄膜在波长575 nm 附近的光学对比度分别为50.4%、69.0%和44.7%。研究表明,在共轭桥联基团上引入吸电子基F原子使得电致变色材料的工作电压增大,并对其电致变色性能具有一定的影响。3种新型Fe(Ⅱ)配位聚合物电致变色材料在水系电解质体系中表现出优异的电致变色性能,对环境友好型电致变色材料的设计及器件的制备具有重要意义。
