分子模拟探究EO-PO共聚物影响添加剂界面膜的机制
2023-12-21龚莹迎
龚莹迎 吕 涯
(华东理工大学化工学院 上海 200237)
由于润滑油应用场合的不同,需要添加多种添加剂以满足油品多项性能指标。润滑油中不同添加剂分子之间,可以各司其职、相安无事,也可能产生协同作用,但也存在相互产生负面干扰的可能性,所以在实际应用过程中润滑油配方筛选十分复杂,往往需耗费大量时间和费用[1-2]。虽然实验研究方法可以相对宏观对比分子添加剂作用及用量对性能指标的影响,但很难从微观上解释添加剂作用的原理及分子之间的相互作用关系。分子动力学(Molecuar Dynamics,MD)模拟技术能够从原子或分子角度解释实验现象,不仅可以有效弥补实验方法研究中的不足,而且它还可以作为一种预测实验结果的手段,为实验提供新的研究思路[3-5]。近年来,分子模拟技术开始应用于研究润滑油添加剂及不同添加剂之间的相互作用。TSAGKAROPOULOU等[6]通过MD的方法研究了甘油单油酸酯(GMO)和聚异丁基琥珀酰亚胺-多胺(PIBSA-PAM)在角鲨烷中的结构和摩擦性能,发现胶束和吸附表面活性剂的存在与较低的动摩擦因数有关,并存在一个最佳的减少摩擦的组成范围。胡雪莹和吕涯[7]通过MD的方法研究了胺类和酚类抗氧剂取代基变化对其抗氧化活性的影响,得出取代基对分子的抗氧化活性有显著影响、且取代基碳数越多抗氧剂分子越活泼的结论。
水的润滑性较差,并且容易引起腐蚀,因此润滑系统应尽量避免含水。然而,通常润滑系统不能绝对防止水渗入。大多数工业油(包括液压油、齿轮油、汽轮机油、压缩机油)和内燃机油都需要良好或出色的抗乳性能,将水污染物与润滑油分离,沉到油箱底部,通过放水阀从润滑系统中分离出去。水分离性能差的润滑油可以形成相对稳定的油包水乳液,极大劣化润滑性能。除了原本基础油所含杂质和使用过程中产生的氧化产物,清净剂、缓蚀剂等具有表面活性的功能添加剂也对润滑油的抗乳化性能产生负面影响。另外一方面,破乳剂是表面活性剂,在特定情况下,可以有乳化作用。这给润滑油配方研究带来很多困难[8]。使用破乳剂是处理采油工业中乳化沥青的重要方法,目前研究人员采用MD模拟方法在乳化沥青破乳剂筛选和破乳机制研究方面进行了有益的探索。NIU等[9]采用MD模拟方法解释了沥青质模型化合物C5Pe分子以C5Pe聚集体的形式吸附在油-水界面上,而EO-PO(环氧乙烷-环氧丙烷)共聚物分子可以取代部分吸附的C5Pe分子,并且可以在界面上形成新的表面屏障,抑制C5Pe分子的吸附。这表明了运用MD分子模拟研究润滑油抗乳化性能和破乳剂机制的可行性。
聚醚类非离子表面活性剂是一种非常重要的表面活性剂,具有优良的表面活性。其中EO-PO嵌段聚醚是环氧乙烷(EO)和环氧丙烷(PO)分别形成均聚长链并在主链上间隔排列的聚醚,其中EO链段可与水作用形成氢键而易溶于水,具有亲水性,而PO链段不溶于水受到水分子的排斥,具有憎水性,这样的结构使得聚醚具有两亲性。当EO-PO聚醚处于油水混合的乳液中时,由于乳液状态不稳定,它将附着于乳液的界面,降低了液体的界面张力,发挥破乳的作用,其性能与EO/PO的比例、EO与PO的聚合方式、分子量、起始剂、封端剂有关,结构有着丰富的可设计性。文中选取的EO-PO共聚物被建模为具有三臂的星形结构,以EO占总质量百分比35%的甘油为基础化合物。
本文作者采用全原子分子动力学模拟方法研究了正辛烷、水共存时各类添加剂分子在界面的乳化效果及添加剂和破乳剂共存时破乳剂的破乳效果。以破乳剂EO-PO、抗氧剂DPA、极压抗磨剂PN、极压抗磨剂S4、缓蚀剂T561为研究对象,分别建立油/水、油/DPA/水、油/PN/水、油/S4/水、油/T561/水、油/DPA/EO-PO/水、油/PN/EO-PO/水、油/S4/EO-PO/水、油/T561/EO-PO/水9个不同的体系,并依次对模拟体系进行能量最小化、结构优化、退火减少分子重叠构象以及各系综条件下的弛豫,直至体系能量收敛到达稳定状态,进而对模拟后的界面形态及各相密度分布、界面膜厚度、径向分布函数、氢键能量等参数来表征添加剂分子的吸附行为,探讨在油水界面上不同添加剂的乳化效果及EO-PO分子协同时的破乳效果。
1 模拟方法
1.1 分子力场
分子力场是描述各种形式的相互作用力对分子势能影响的势能函数。文中用于模拟的软件是 Materials Studio 2019(MS)版本[10],它包括CVFF、PCFF、COMPASS、COMPASS Ⅱ以及通用力场等。其中COMPASS 力场是第一个提供大范围内模拟与预测单个粒子以及凝聚态物质的相关物理性质与几何结构的力场。该力场涉及到的很多参数都是以量子力学为基础获取的。COMPASS力场与其他力场的关键区别在于,力场中的非耦合相互作用势函数与力常数不是简单地通过结合单粒子系统的数据产生的,而是通过对液体与晶体进行 MD 模拟优化形成的,其精确度更高,尤其振动频率方面。COMPASS力场的适用范围为小分子和聚合物等体系,而且力场参数不因研究对象或模型的不同而变化。该力场对不同界面以及材料模拟较为精确,经常被用于表面活性剂在界面的吸附行为分子模拟。COMPASS II力场是COMPASS力场的重要扩展,COMPASS II 扩展了 COMPASS 的现有覆盖范围,更新现有参数或添加其他功能组的参数,当前版本的直接前身被保留并可供使用,以供验证和延续现有项目。
1.2 周期性边界条件
周期性边界条件是为了在有限的空间内尽可能准确地代表所研究的真实系统。其基本原理是假设要建模的系统被无数个相同单元所包围,充满整个空间,并且每个单元具有相同的粒子分布和速度。由于这种周期性排布,一个粒子从单元的一侧离开后,必须有一个等效粒子从单元的另一侧进入,从而保持单元中粒子的数量不变。因此模拟计算不需要计算一个无限系统,只需要处理和存储一个小单元的数据,并消除了试图减小粒子数量所造成的有限尺寸效应。模拟模型包括有5种不同形状的周期系统:立方格子或者平行六面体、六方柱体、无顶角八面体、菱形十二面体和延长的十二面体。在Materials Studio 2019(MS)中 Amorphous Cell 模块建立盒子使用的是立方体或者平行六面体。
1.3 模型构建
所有计算采用Materials Studio 2019(MS)软件包进行计算。图1所示分别为油分子、水分子、EO-PO、DPA、PN、S4、T561的构型示意图。首先通过Visualizer模块分别构建单个油分子、水分子、EO-PO分子、添加剂分子模型,并采用Forcite模块中Geometry Optimization工具对分子进行结构优化,使分子为最优结构;其次,利用Amorphous cell模块中Construction工具分别构建对应分子层,初始盒子大小为2.89 nm×2.89 nm×2.89 nm,其中油相包含127个正辛烷分子,水相包含800个水分子,EO-PO分子层和添加剂分子层分别包含9个对应的分子;最后在Visualizer模块中build layer工具构建油/水、油/DPA/水、油/PN/水、油/S4/水、油/T561/水、油/DPA/EO-PO/水、油/PN/EO-PO/水、油/S4/EO-PO/水、油/T561/EO-PO/水9个不同的体系,其中油/水体系用来作为空白对照,其他分别为加入了不同添加剂及在此基础上加上等量的EO-PO分子[11]。
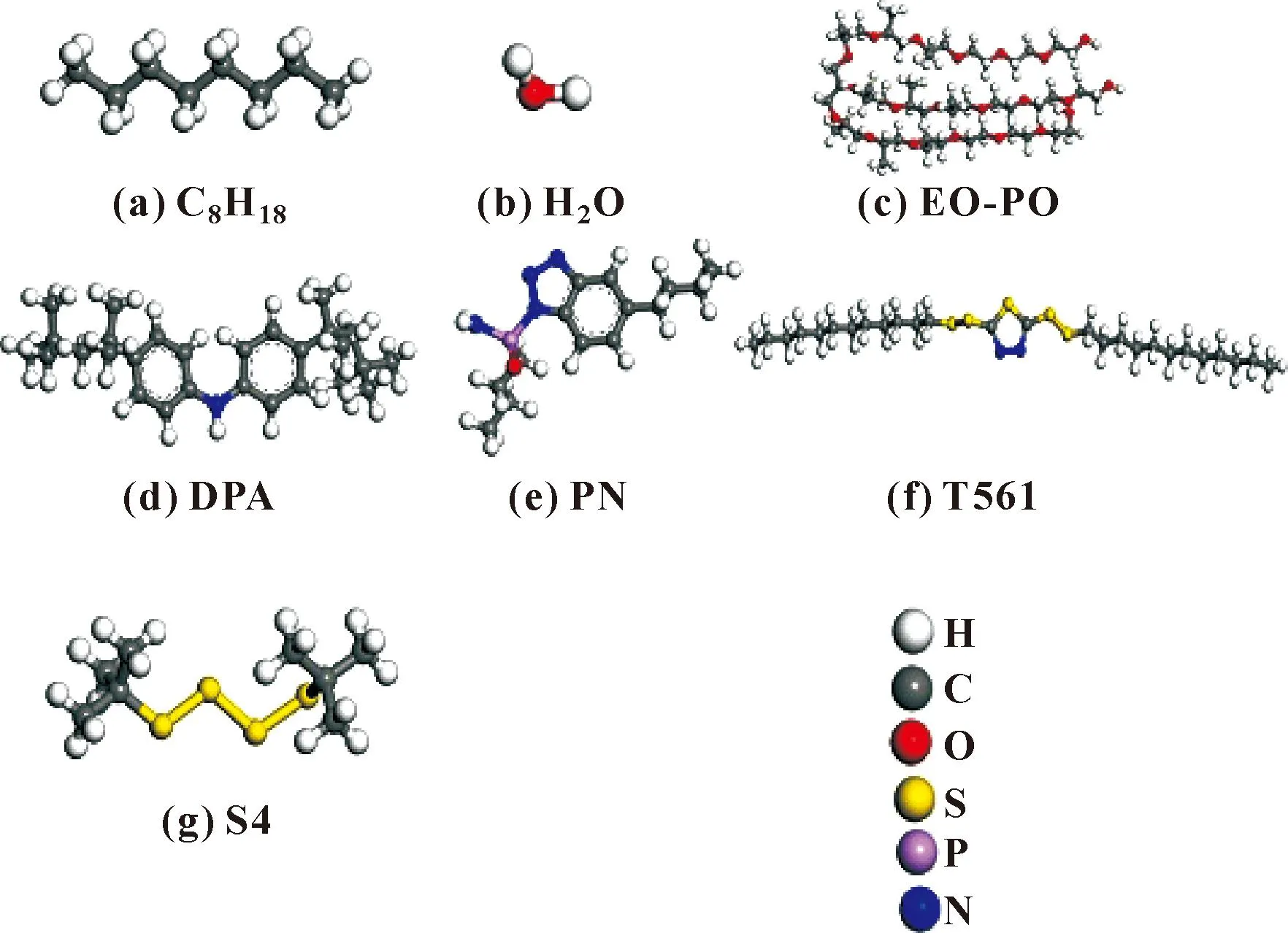
图1 C8H18/H2O/EO-PO/DPA/PN/T561/S4分子模型
1.4 模拟细节
模拟过程在Forcite模块,过程中全程采用CompassII力场,截断半径1.25 nm,粒子间长程库仑静电作用的计算采用Ewald算法,粒子间Vdw采用Atom based算法,动力学弛豫过程中温度设置为298 K,选择常压即0.1 MPa为模拟时的压力,模拟步长为1 fs,并以5 ps的时间间隔收集轨迹[12]。具体模拟步骤如下:
(1)对模拟体系采用 Energy进行能量最小化消除高能构象;
(2)将体系进行时长为 2 000步的退火处理,以去除分子重叠和减少分子间内应力的影响,退火过程中温度先从300 K升至500 K最后再降至300 K,共进行5次循环,每个循环坡道加热温度间隔为20 K;
(3)将优化完备后的体系放在NVT系综下进行100 ps的动力学弛豫,以保持体系的外压平衡,初始速度选项设置为Random,以使体系在NVT系综下产生初速度,温度控制选取Andersen方法,直至体系达到稳定状态,平衡稳定的标准为体系中的温度和能量的波动范围在5%以内;
(4)将体系放在NPT系综下弛豫400 ps,以确保体系中各组分密度能最终达到合理数值,初始速度选项设置为Current,承接体系在NVT系综下持有的速度,温度控制选取Andersen方法,压力控制采用Berendsen方法,直至体系平稳,收敛标准为体系的温度、能量、密度以及盒子大小在5%以内波动;
(5)将体系置于NVT系综下弛豫500 ps,待温度波动范围在5%以内体系达到平稳状态后,进行数据收集统计。
2 结果与讨论
2.1 界面形态
各体系动力学弛豫过程结束(1 000 ps)形态如图2所示。这里需要特别强调的是,随着模拟的进行会有少部分油分子从体系的上侧移出并从下侧进入,这是由于周期性边界条件所造成的,并不会对模拟结果产生影响。在初始状态下,各组分分子均匀分布,随模拟的进行,各组分分子秩序变得混乱,并发生了不同的变化,从图中可以看出:(a)在未添加EO-PO分子的体系中(见图(A1)、(B1)、(C1)、(D1)),添加剂分子层连接油相和水相,聚集在油水混合界面上,其中部分添加剂分子会溶解于油相中,而绝大部分聚集在油水界面上,形成对应的界面膜;(b)在含EO-PO分子的体系中(见图(A2)、(B2)、(C2)、(D2)),添加剂分子和EO-PO分子将油相和水相完全分隔开,油水混合界面消失,其中EO-PO分子部分偏向于水相中,部分偏向于油相中。

图2 各体系模拟动力学弛豫1 000 ps结果
2.2 密度分布和界面厚度
通过计算体系在x、y、z方向的密度分布,可得到各方向的密度分布图,密度分布图能够可视化显示出体系的各组分在体系中某处含量的多少。相对密度(Relative Concentration)表示组分在一定厚度区间中的密度与其在体系中密度(平均密度)的比值,使用MS中Forcite的Analysis中Relative Concentration功能分析得到各组分沿Z轴方向的相对密度分布[13]。
图3所示为各体系平衡时沿Z轴密度分布情况。从图3(a)可以看出,在油/水体系中经过模拟平衡后水的密度值为0.99 g/cm3,油的密度值为0.72 g/cm3[14],结果接近于国际温标下水的密度和正辛烷的密度,满足利用分子模拟手段解释研究对象机制性质所需的精度要求,故认为所采取的计算方法具有科学合理性。在油/水体系中,经分子动力学模拟,油分子和水分子沿Z轴小幅运动,并在油水之间形成一层很薄的膜,薄膜厚度约为0.44 nm[15]。正是因为薄膜的存在,这些界面膜给液滴聚并造成不同程度的动力障碍,使得水以小水滴的形式存在于油相中,难以聚集在一起,让油水分离变得困难,从而降低了油品的性能。

图3 不同组分沿Z轴方向密度分布
从图3(b)、(d)、(f)、(h)可以看出,当在油水体系中分别加入DPA、PN、S4、T561添加剂分子时,添加剂分子均在界面呈单层膜分布,界面区内油/水均表现出明显的密度下降,添加剂组分的密度分布曲线与油相密度分布曲线交叉所形成区域的面积,明显大于其与水相密度分布曲线所形成的区域面积。这是因为这4种添加剂分子含有的亲油的烷基链会与油相分子间存在范德华作用力,与其发生一定程度的互溶,同时添加剂分子与水分子之间还存在静电作用和氢键作用,使水分子聚集在周边[16-17]。这2种作用方式共同促进了界面膜的形成。然而界面膜的存在使得水相和油相的过渡区域变大,油水混合部分之间的排斥力减弱,使得油相中水滴难以聚集在一起,油水乳状液更加稳定存在,油水分离相对于单纯的油/水体系会更加困难。
从图3(c)、(e)、(g)、(i)可以看出,在含有各添加剂分子体系的基础上,加入等量的破乳剂EO-PO分子时,油相和水相的交叉区域消失,油相和水相分成明显的两相,EO-PO分子和添加剂分子位于油相和水相之间,说明EO-PO分子可以实现油水的分离,具有一定的破乳效果。这是因为EO-PO分子中亲水链段EO比添加剂分子的亲水能力更强,从而可以顶替在油水界面处聚集的添加剂分子,破坏添加剂分子所形成的界面膜。以DPA分子为例,如图4所示为DPA分子、EO-PO中亲水基团和疏水基团在界面的相对浓度分布,发现随着时间的推移(从模拟过程开始到1 000 ps),DPA分子由界面逐渐向油相移动,而EO基团逐渐向水相靠近,表明EO-PO分子的界面活性更高。

图4 DPA分子/EO/PO在界面上沿Z轴方向相对浓度分布
体系的界面厚度可以直观地反映出添加剂分子在界面吸附程度的强弱,是考察界面性质的一个重要指标[18]。基于上述界面密度分布曲线可以计算出界面厚度,其中,界面区分为水区、油区以及总区,界面油区及水区厚度的计算选取“10%~90%”标准、总区厚度采用“90%~90%”标准 (水层密度的 90%到油层密度 90%之间的区域厚度)[14],各体系界面厚度见表1。
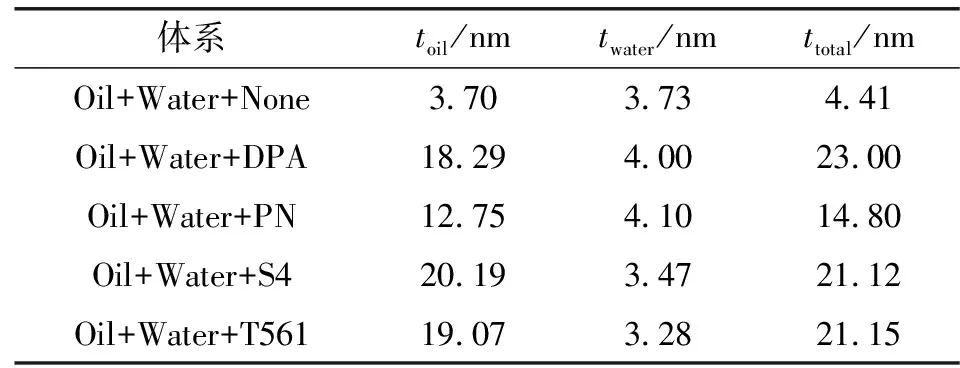
表1 不同体系的界面厚度
对于含有EO-PO分子体系而言,由弛豫状态(见图2)和密度分布(见图3)可看出,油相和水相没有混合在一起,故不存在油水界面,因而界面厚度的定义不再适用。而对于其他体系而言,油水之间存在一定的混合,其总界面厚度大小表明界面区添加剂分子间的渗透性能,界面厚度值越大,界面膜稳定性越好。由表1可见:①较于纯油水体系,加入不同添加剂分子后,各界面区厚度呈现不同幅度的增大趋势,总界面区厚度DPA最大,S4、T561次之(两者厚度大致相等),PN最小,说明DPA、S4和T561分子体系形成的界面较为粗糙,其在界面区吸附效果佳,较其他体系形成界面膜时更为稳定;②油相和添加剂分子之间形成的单一界面厚度要显著大于水相和添加剂分子之间形成的厚度,二者厚度由于添加剂分子渗透效果不同呈现出3~6倍的数量关系,其中twater相差不大,造成二者厚度差异主要是因为作为添加剂分子的烷烃链会部分溶解于油相,使得界面变得相对模糊,这也与前面对其界面形态分析的结果相印证。
2.3 径向分布函数
径向分布函数(Radial Distribution Function,RDF)是描述体系中目标粒子在距离某一参考粒子指定距离时出现的概率函数[19],可以用来分析体系的微观结构,通常将其定义为与 A类中心原子距离为r的球壳中出现 B类原子的概率,该函数可以反映出两原子之间的相互作用,峰值的位置对应 A-B间相互作用的最可能距离。RDF值越高代表分子间在r距离内出现的概率越高,分子间的作用力越强。径向分布函数公式见公式(1)。
g(r)=dN/(4πρr2)
(1)
dN为指定粒子A在距离为r→r+dr范围内与另一被指定粒子B的数目;ρ为指定粒子B的平均数目密度,计算方法为:用所研究粒子总数目除以体系总体积得出。
图5(a)描述了在EO-PO分子加入前后各添加剂分子与水的RDF,结果表明,随着各添加剂分子与水的分离度的增加,二者之间的g(r)单调增加。对于同时含有EO-PO和添加剂分子的体系,添加剂分子在无EO-PO分子的体系中与水的相互作用力更强,说明EO-PO分子的加入,使得各添加剂分子在界面活性降低,其中PN和DPA分子下降程度较为明显。

图5 添加剂分子和EO-PO分子与水的RDF
图5(b)描述了EO-PO分子中亲水基团EO中氧原子与水中氧原子的RDF,结果表明,在0.26和0.53 nm处分别出现一个尖锐的峰[20],主峰对应的位置均在0.26 nm处,则EO-PO分子在此处与水相结合最紧密,这是EO链段和水分子之间形成强烈的氢键作用所致,此时出现第一水化层。在主峰位置后面0.53 nm处出现一个峰值较小的次峰,峰值偏低,这应该是远程作用力作用导致的物理水化层,此时出现第二水化层。通过比较峰值发现,在含有不同添加剂体系中,EO-PO分子与水作用力大小为 PN最大,DPA次之,S4、T561最小(两者大致相同),说明EO-PO分子在含PN和DPA分子体系发挥破乳的作用较好。
2.4 氢键能
氢键是分子间作用力的一种,氢原子通过共价键与电负性大的X原子键合,如果它们靠近电负性大半径小的原子Y(O、S、N等),X和Y之间形成一种特殊的X-H…Y形式的分子间或分子内相互作用,是一种永久偶极之间的作用力。由于EO-PO分子和各添加剂分子中含O、S、N 原子,可以与水分子之间形成O-H…O、O-H…S、O-H…N、N-H…N 等氢键,为解释EO-PO分子和各添加剂分子在油-水界面上的聚集行为及吸附效果,使用Forcite中Energy模块Dreiding force field计算了各体系的氢键能,结果如图6所示。
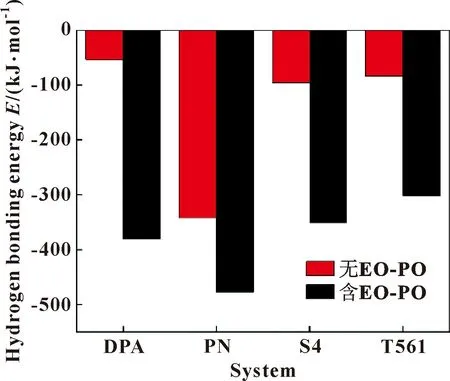
图6 各添加剂体系加入EO-PO分子前后的氢键能
氢键能在此力场下显示为负值,其绝对值越大,则说明EO-PO和添加剂分子与水分子之间的作用力越大,界面活性越高,对应体系越不稳定[21]。结果表明:①未加入EO-PO分子前,添加剂分子均和水发生了不同的相互作用,其中添加剂分子与水之间作用力大小为PN最大,S4、T561次之,DPA最小,这与上述界面厚度的结果相一致;②加入EO-PO分子后,由于EO-PO分子比添加剂分子具有更强的亲水性,会顶替添加剂分子与水之间的相互作用,此时EO-PO和添加剂分子与水之间的相互作用合力大小为PN最大,DPA次之,S4、T561最小,表明在含有PN和DPA分子的体系中,EO-PO分子对其界面膜破坏较大,破乳效果较佳,这与上述径向分布函数的结论相一致。
3 结论
通过分子动力学模拟考察破乳剂EO-PO分子及DPA、PN、S4、T561添加剂分子在正辛烷/水界面上对界面性质的影响,探究各体系动力学弛豫界面形态、密度分布和界面厚度,以及各体系RDF和氢键能。主要结论如下:
(1)各添加剂分子均会在油水界面形成对应的界面膜,使得油水混合乳状液相较于单一的油水体系更为稳定,界面稳定性排序为DPA最稳定,S4、T561次之,PN稳定性最差;而EO-PO分子的加入,由于较添加剂分子具有更强的亲水性,可以在界面上顶替添加剂分子与水相互作用,从而达到破坏界面膜的稳定性,使得油相和水相完全分开的效果,具有一定的破乳作用。
(2)在含有不同添加剂的各体系中,EO-PO分子与水有不同程度的相互作用,其中在含有PN和DPA分子的体系中,EO-PO分子对其界面膜破坏较大,破乳效果较佳。
