电合成过氧化氢电催化剂的设计及进展
2023-12-16谢东升宋洋丁莹王耀彬沈钰凡赵云霞
谢东升 宋洋 丁莹 王耀彬 沈钰凡 赵云霞
电合成;过氧化氢;两电子氧还原反应;催化机制;活性位点
0 引言
过氧化氢(H2O2)是一种高效、绿色氧化剂,广泛应用于医药、化工、环保及纺织等行业.此外,过氧化氢可以作为晶圆清洁剂和制环氧丙烷(HPPO)工艺的环氧化试剂,在半导体行业迅速发展及全球公共卫生安全等问题频发的背景下,需求量急剧增长[1-3].据报道,2015年全球H2O2产能已达550万t,预计2022年可达650万t[4],而现有的生产规模仍不能满足未来需求[5-8].
传统工业大多采用间接能耗大、能源密集型的蒽醌法生产H2O2,由Riedl[9]于1939年首次开发.该工艺需要使用昂贵的钯催化剂,通过蒽醌的连续氢化和氧化制备H2O2.然而,该方法存在诸多缺陷,例如,同时使用氢气和氧气导致该工艺在气体运输和储存方面存在安全隐患,并且需要额外的蒸馏提纯和分离步骤保证其质量分数达70%,以降低运输成本[10-11].因此,开发低成本、高效及分散式生产工艺,成为研究热点.近年来,使用氢气(H2)和氧气(O2)的分布式生产H2O2工艺成为焦点,其使用的钯-锡催化剂对H2O2的催化选择性高于95%[12-14].但在一定浓度下,H2和O2混合易燃易爆,H2需要大量的其他稳定气体(如N2、CO2)进行稀释后才可以使用,因此该工艺不适合大规模商用[15].
研究发现,电催化氧还原(ORR)生产H2O2极具商用价值,如图1a所示.ORR活性电催化剂主要应用在需要高效的4e-传输机制的燃料电池,而研究人员发现,开发具有2e-传输机制的ORR催化剂生成H2O2的过程成本低且能耗小.在酸性水溶液中,4e-和2e-ORR的两种途径分别为

图1 (a) 2015—2019年对2e-ORR和4e-ORR研究报道数量趋势图(以2015年为基准);(b) 理论计算ORR过程火山图,使用OH*或OOH*的结合能作为描述的路径,产物为H2O(蓝色)和H2O2(红色)[16]Fig.1 (a) Trend charts of the number of published reports on 2e--ORR and 4e--ORR in recent years;(b) volcano plot for theoretical calculation of the ORR process,using the binding energy of OH* or OOH*as the described path,and the products are H2O (blue) and H2O2 (red)[16]
4e-ORR:

(1)
2e-ORR:
(2)
(2a)
(2b)
E0是根据反应的自由能计算出的标准平衡电位,将其转化为相对可逆氢电极电位(VRHE).为使反应热力学不受pH的影响,通常在300 K下通过式(3)转换:
E(VS,RHE)=ERef+0.059×pH+Etest,
(3)
在碱性溶液中,质子从水中获得,则两个反应变为
4e-ORR:

(4)
2e-ORR:
(5)
(6)
式(6)是pH大于11.7时反应的变化,对于2e-途径的ORR来说(式(2)、(5)和(6)),这种变化的关键是对OOH*物种的结合强度[16].
Kulkarni等[17]和Peterson 等[18]使用含氧物种的吸附能ΔGOOH*或ΔGOH*预测了氧还原反应的结合能强弱(图1b).在火山图中,对OOH*的结合强度决定了反应路径向火山顶峰左(4e-途径形成水)或右(2e-途径形成过氧化氢)的偏移,从而解释了4e-和2e-的竞争反应.峰值左侧表示较强的OH*结合点,OH*形成H2O的自由能减小,表示4e-ORR的选择性大于2e-.在火山峰的顶端,H2O2和H2O的形成都有很高的活性,表明4e-和2e-氧还原反应同时发生.而从两电子火山峰向右移动,两图相互重叠,无论是通过OOH*的化学解离还是电化学还原,都较难打破O—O键,增加了H2O2选择性,但也同时减弱了O2对OOH*的活化.
由于选择性地生成H2O2需要调整氧还原反应的反应路径,因此催化剂的选择性和反应活性成为筛选的关键.Adzic等[19]首次在Au(111)和Au(110)的表面观察到了氧还原反应通过两电子途径发生.此后研究人员开始关注其他金属材料电子转移路径的调整策略,以寻求通过平衡OOH*的结合能力和催化活性,获得同时提高H2O2选择性和产率的理想催化剂.例如一系列贵金属及其合金(Au-Pd[20-21]、Pd-Hg[22]和Au[23])被证明具有较低的过电位和较高的H2O2选择性(>98%).目前,铂族金属材料(PGM)被认为是最先进的ORR催化剂.通过一系列分散铂族催化剂反应位点的调整策略,使反应途径发生了从四电子到两电子的转变,包括现场隔离形成单原子、表面涂覆非晶态碳层和制造包覆壳结构等方法.但是,贵金属的稀缺性导致其原料成本较高,阻碍其大规模商用和工业替代的工艺开发.最近,地球储量较为丰富的碳基材料成为研究热点,其具有灵活的精细结构及优异的电化学稳定性等特点,被认为最有潜力替代贵金属催化剂[24-26].在早期对2e-ORR过程的研究中,原始催化剂的电子结构对反应中间产物(OOH*)解吸能力不理想,导致对H2O2的选择性较低.为弥补这一点,有很多研究采用界面工程和动力学的手段优化催化剂表面官能团,促使生成的过氧化物及时从表面脱离,避免被进一步还原.
鉴于此,本文阶段性综述了4e-ORR催化剂通过结构调节与掺杂等方法使反应过程转变为2e-路径的调整策略,这些策略的报道有助于进一步开发低成本、高选择性的先进催化材料,以实现高效生产H2O2.最后,从高效电催化剂的合理设计、反应工程、电化学反应器的设计等方面阐述了电化学合成H2O2未来发展面临的主要挑战、机遇及其潜在的应用前景.
1 金属-碳基电催化剂
1.1 几何效应-碳偶联单原子催化剂
单原子催化剂(SACs)由于其打破传统贵金属尺度效应关系,具备同时获得高活性和H2O2高选择性的特点,成为近期研究热点.氧的吸附存在“端对吸附”(O2分子为垂直取向)和“侧对吸附”(O2分子为平行取向),单原子催化剂的优异特性在于活性中心被原子隔离,在这种结构下O2在催化剂表面呈端对型(图2a),导致了O—O键反应势垒增大,O—O键断裂难度增加,进而使SACs易于通过2e-ORR途径产生更多的H2O2[27-32].
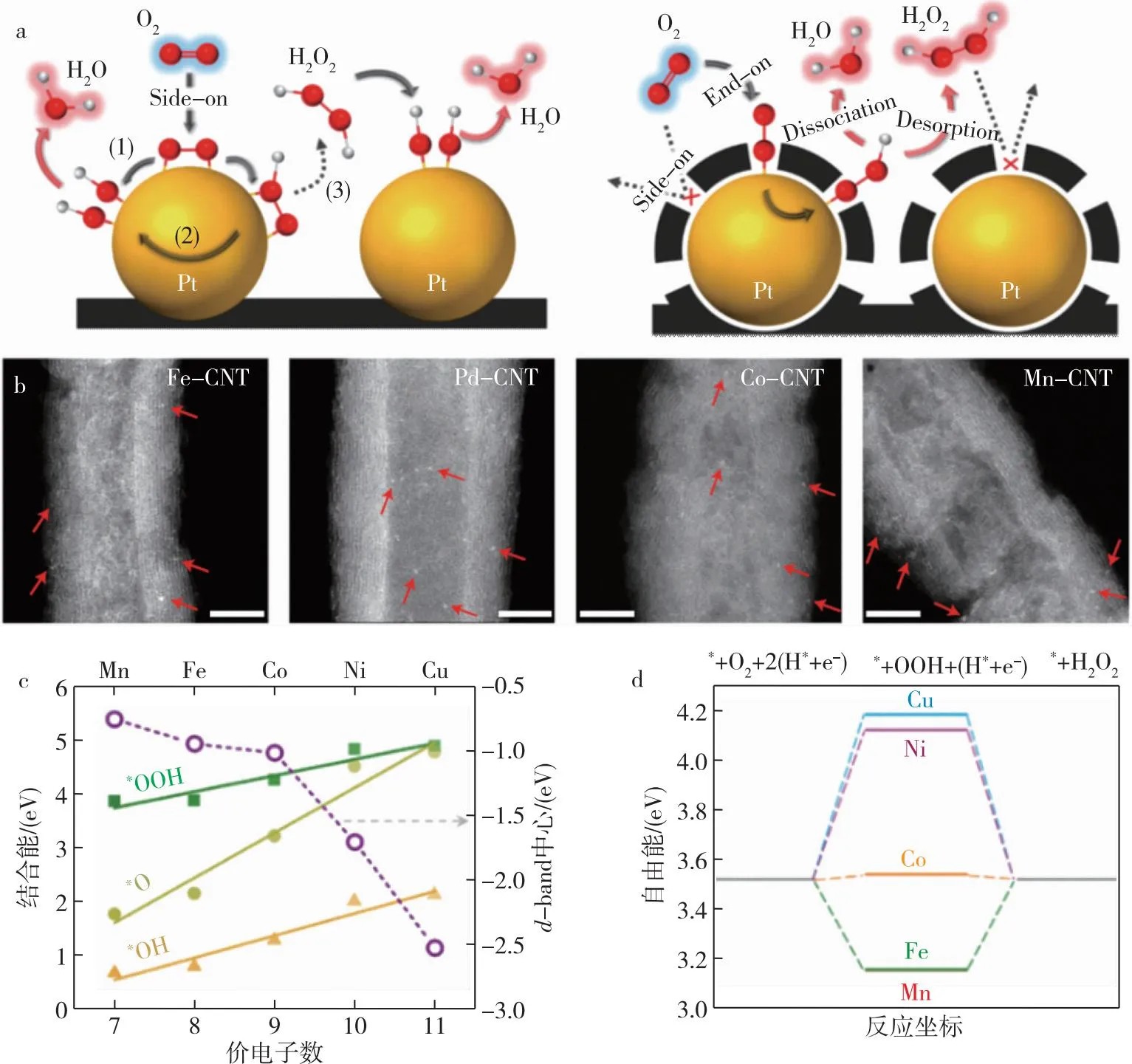
图2 (a) 原始Pt/C和Pt涂覆的无定形碳层[31];(b) M-CNT催化剂的TEM表征图,HAADF-STEM中的亮点(用红色箭头标记)代表一些典型的金属单原子[33];(c) OOH*、O*和OH*在M-SAC上的结合能(M=Mn、Fe、Co、Ni和Cu)和M-SAC中M原子的d-band中心[34];(d) 在U=0.7 V时,SAC上2e-ORR的自由能图[34]Fig.2 (a) Pristine Pt/C and Pt coated by amorphous carbon layer[31];(b) TEM representation of M-CNT catalyst,bright dots in HAADF-STEM (marked by red arrows) represent some typical metal monatoms[33];(c) binding energy of OOH*,O* and OH* on M-SAC (M=Mn,Fe,Co,Ni,and Cu) and d-band center of M atom in M-SAC[34];(d) free energy diagram of 2e--ORR on SAC at U=0.7 V[34]
研究表明,单原子金属的负载量和粒子间距是影响H2O2选择性的直接参数,若单个位点的金属催化剂纳米粒子尺寸减小到一定水平或分布较稀疏,电子转移路径会向两电子方向转变,H2O2会成为O2还原的主要产物[4].而粒径较小的金属纳米颗粒对氧分子具有较低的结合能,有利于提高ORR两电子途径中OOH*的生成[33].
Song等[26]报道了一种石墨烯支撑的Pt单原子(质量分数为0.48 %)催化剂,在0.8 VRHE下电流密度约为3.10 A·mg-1,为商用Pt/C催化剂的57倍.他们发现平均大小为1.02~0.02 nm的单个Pt位点都可能成为独立的活性位点,并且进一步研究发现ORR反应路径与Pt物种的粒径大小直接相关[25-26],随着Pt尺寸减小到原子水平,没有可用的相邻位点来破坏孤立Pt原子连接的O—O键,ORR过程向两个电子路径方向进行.旋转环盘电极(RRDE)结果表明分散的Pt原子对H2O2选择性高达95%.除此之外,提高OOH*中间体的吸附能,也是提高过氧化氢生成率的另一个有效途径.
早期有研究者报道,金属-氮共修饰碳(M-N-C,M为过渡金属)的结构设计和配位调控是提高催化活性的有效策略.Gao等[34]结合密度泛函理论计算(DFT),通过对氮掺杂石墨烯(NC)中M(M=Mn、Fe、Co、Ni、Cu)单原子催化剂ORR性能比对(图2c),发现M原子的d-band中心相对于费米能级从Mn向Cu的能量下移[35-36],Co-SAC在U=0.7 V下具有最佳d-band中心,ΔGOOH*=3.54 eV几乎位于火山图的顶点,在0.6 V的电位下,Co-NC的电流密度达到1 mA·cm-2,具有高活性、高选择性,而对H2O2选择性也超过了90%.
近日,类似卟啉配体金属中心的多项单原子催化剂M-N4(M为过渡金属)-SACs被认为在氧还原反应中具有高度的活性[37-42],但其2e-ORR的反应机理存在争议[43-44].Jung等[16]合成了由掺氮石墨烯支持的Co-N4单原子催化剂,他们通过调整官能团O*使其吸附在Co-N4附近,如图2d所示,ΔGOOH*从3.9 eV增长到4.1 eV.这种由C—O—C环氧化物包裹的Co-N4,其电流密度在0.65 V下达到了2.8±0.2 mA·cm-2,在Co-NG(O)负载量约为1 mg·cm-2的情况下,H2O2产率高达418±19 mmol·g-1·h-1[30].
Wang等[45]研究了包括Fe、Pd、Co和Mn等一系列过渡金属(TM)的单原子配位基序,通过浸渍还原法将TM单原子固定在碳纳米管(CNT)的空位上(图2b).研究发现在0.1 M(M指mol/L) KOH碱性条件下,Fe-CNT在0.822 V达到了起始电位并形成了0.1 mA·cm-2的电流,在RRDE测试中H2O2的选择性也达到了95%以上.他们又进一步研究了N和O对反应路径的调节机理,发现氧气在GDL电极的促进下,使得Fe-CNT-O拥有了更低的起始电位0.76 V,H2O2的生成速率也达到了43 mA·cm-2.最后通过密度泛函理论(DFT)计算证明发现,反应的两种产物H2O2(2e-)和H2O(4e-)路径分别由M—C—O基序中C和Fe活性位点控制.
1.2 协同效应-碳偶联金属氧化物催化剂
除了单个金属原子形成的纳米颗粒可以改良碳基材料的催化活性之外,过渡金属氧化物(MnO2、Fe3O4等)作为ORR催化剂研究最广泛的化合物之一,因为其易开发、含量丰富等特点,常常与高导电的碳载体形成复合材料,用于提高ORR性能.在二电子途径的选择上,先前已有报道证明,Fe3O4催化剂在2e-ORR产生H2O2的活性位点主要位于Fe(II)表面[46].Barros等[47]以NaBH4为还原剂采用沉淀法成功合成了比表面积为450 m2·g-1的Fe3O4/石墨烯催化剂;他们发现在0.3 VSCE的情况下,该复合材料的电流密度达到1.12 mA·cm-2,高于石墨烯(0.85 mA·cm-2),体现了Fe3O4与石墨烯的协同作用,最终在0.2~0.7 VSCE的电势范围内,H2O2的电合成选择性大于60%.Xiao等[48]利用电化学和单分子荧光显微镜(EC-SMFM)的方法研究了单个Fe3O4纳米粒子2e-ORR的催化动力学(图3a),揭示了2e-ORR过程中催化活性的等速关系,认为单个纳米粒子间存在动态不均匀性并且存在补偿效应.

图3 (a) Fe3O4 NPs单粒子2e-ORR过程的简图[48];(b)Nb2O5高度分散在rGO中的TEM、HRTEM和SAED图像[49]Fig.3 (a) Schematic of the 2e- ORR process of Fe3O4 NPs[48];(b) TEM,HRTEM and SAED images of Nb2O5 highly dispersed in rGO[49]
Carneiro等[49]采用水热法在还原性氧化石墨烯(rGO)薄膜上制备了Nb2O5纳米颗粒(图3b),他们将还原的石墨烯和原位生成的Nb2O5复合形成纳米复合材料并与炭黑做出对比.他们发现Nb2O5-rGO表现出更高的环电流,在酸性条件下H2O2的产率达到了85.3%且峰值电位向正方向移动,进一步证明了Nb2O5纳米颗粒(金属氧化物)与rGO片(碳材料)之间对2e-ORR具有明显的协同作用.
2 非金属基催化剂两电子ORR性能的提升策略
碳材料由于在环境中丰度高、分子结构可调节、电子导电性高及传输稳定性强等特点,近年来在氧还原反应(ORR)电催化中的应用研究快速增长[27,50-52].在先前的研究中碳基材料已被报道是电合成过氧化氢最具前景的电催化剂,通过对碳基材料的掺杂改变对O2的化学吸附特性,能够有效地控制O—O键的断裂[53-54].在此之后,各种不同形式的纯碳材料如石墨烯、碳纳米管和富勒烯以及各种碳混合材料如氮掺杂、硫掺杂、氟化物和氧化碳等催化剂,都被相继报道对2e-途径氧还原反应生产过氧化氢具有较高的催化活性和选择性[55-57].因此对碳基材料2e-ORR催化材料在反应路径策略调整及制备方法等方面的总结,有利于从反应机理的角度,对开发出更高效的H2O2催化剂产生更多重要的启发.
2.1 活性中心缺陷的设计策略
活性中心的基本特征是为特定的ORR过程提供低反应势垒和高极限电位.为了改善碳基材料的结构与活性之间的关系,引入了氧官能团(OFG)以增强两电子路径碳基催化剂的选择性.这种OFG中的氧含量与催化活性呈正相关[58-61].但在对含氧官能团催化剂活性中心位点的筛查过程中,研究者发现,由于氧官能团不可避免地将悬挂键饱和带有复杂的多组分边缘与表面选择性机制混淆,即表面羧基也可以捕捉过氧化物,使用现有分辨率的材料表征和原位拉曼光谱等手段无法区分相似的官能团,导致含氧官能团碳基材料2e-ORR催化活性中心一直存在争议[62-67].
设计结构与官能团的缺陷是研究氧化碳催化活性中心的间接方法.Kim等[58]利用光谱结构表征和原位拉曼光谱等手段研究了多层氧化石墨烯 (F-mrGO),结果表明sp2碳位形成的OOH-物种与环醚基团有直接关联(图4a),在碱性条件下,高浓度的环醚基碳催化剂仅在10 mV过电位下就可以产生H2O2,并且选择性接近100%.这些结果都表明sp2杂化碳附近的环醚缺陷是产生过氧化氢的活性中心,且通过控制醚基缺陷,能够调控ORR对4e-或2e-反应途径的选择.为了进一步研究官能团对碳基材料的影响,Han等[68]利用醚、羧基和醌等功能基团来修饰预活化石墨纳米片(GNP)的悬垂边缘,发现在0.75 V电位下,含喹诺酮类化合物的石墨纳米片(GNPC=O)H2O2产率高达97.8%.通过对醌、羧酸和醚环基团独立分子的氧气还原产过氧化氢 (ORHP)性能比较,发现醌类化合物JH2O2(0.7 vs 0.5 mA·cm-2,在0.65 V电压下)和Tafel斜率均优于其他基团,证实了醌类化合物是ORHP的活性中心.他们通过DFT计算证明,醌基表现出很高的选择性,该理论研究也进一步验证了上述结果.
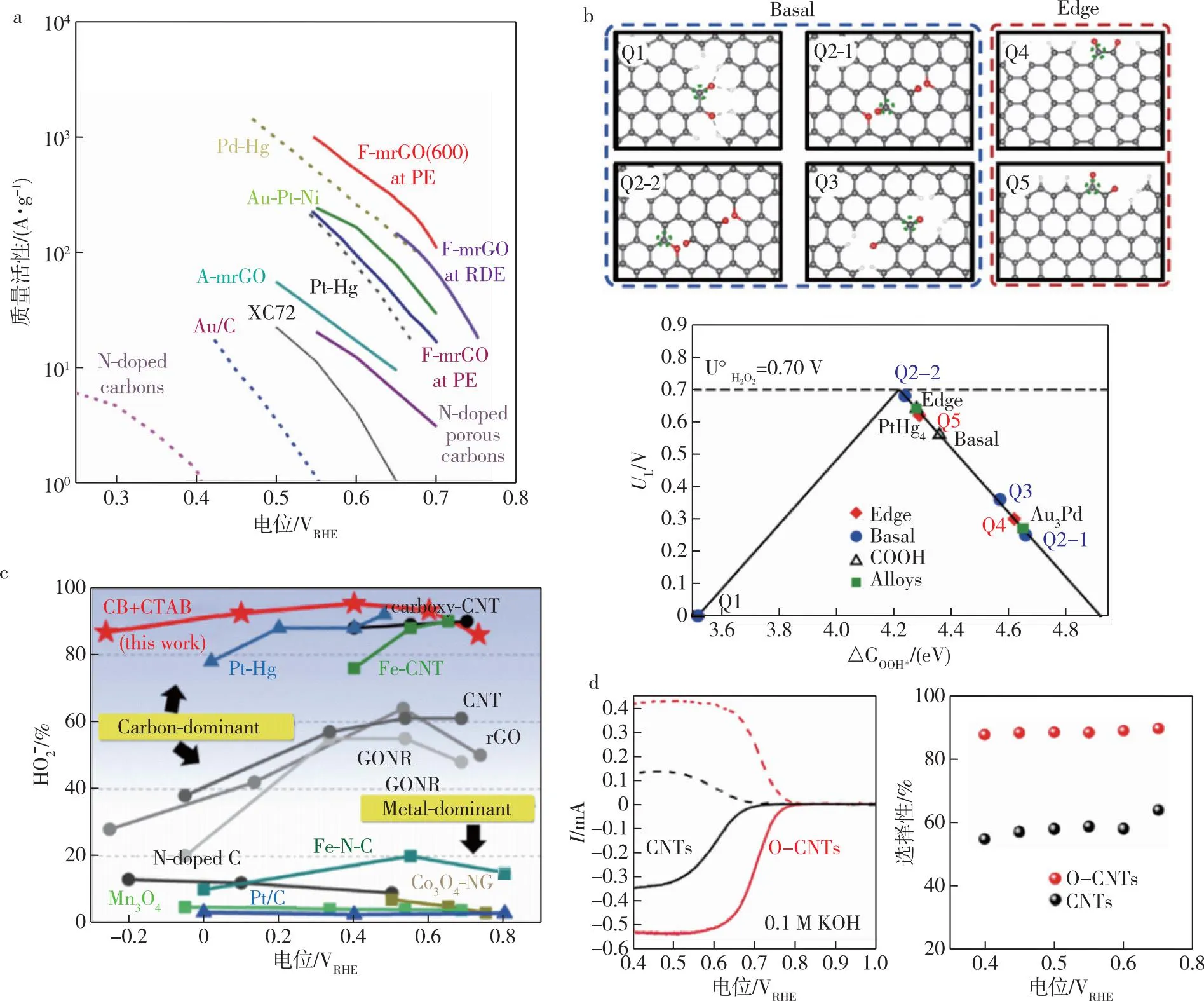
图4 (a) 不同电催化剂生产H2O2的质量活性对比[58];(b) 不同氧化基团的理论分析[68];(c) 电催化剂体系中过氧化物的选择性和潜在的窗口宽度的对比[69];(d) CNTs和O-CNTs的氧还原性能[59]Fig.4 (a) Mass activity of different electrocatalysts for H2O2 production[58];(b) theoretical analysis of different oxidation groups[68];(c) comparison of peroxide selectivity and potential window width among the electrocatalyst systems[69];(d) oxygen reduction performance of CNTs and O-CNTs[59]
同样地,通过对比典型碳材料表面化学结构与其催化氧还原生成过氧化氢的反应活性之间的关联关系,Lu等[66]用不同表面氧官能团的氧化炭黑(OCB)证明了二电子氧还原反应的选择性与碳材料表面的羰基和羧基含量呈线性正相关关系,是产生过氧化氢的主要活性中心.

Wu等[69]按照这一思路利用界面工程的方法和反应动力学来促进炭黑(CB)电极上的2e-ORR反应,他们将阳离子表面活性剂十六烷基三甲基溴化铵(CTAB)作用在CB表面,由于CTAB层的原位库伦作用的驱动,表面过氧化氢一旦生成就被从碳表面拉脱,而羧基在反应中起到易解吸的重要作用(图4b、c).在电位0.8 V的碱性介质中H2O2的选择性高达95%,并且证明了碳的边缘缺陷对过氧化氢的高选择性没有贡献.
鉴于纳米碳/表面活性剂复合电极材料在过氧化氢电合成反应中的优异催化表现,整个反应体系能耗低、绿色、可持续、稳定性好的特点,尤其是对该体系结构-功能关系的深刻理解,这项研究工作对未来设计开发具有实际应用前景的高产率、高稳定性和低成本的电合成过氧化氢化合物体系具有重要的指导意义[66-67,70].
2.2 杂原子掺杂非金属基催化剂
2.2.1 氮掺杂碳及其功能化
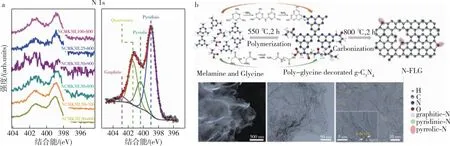
图5 (a) 氮掺杂碳的N 1s核能级区域高分辨率XPS光谱[74];(b) N-FLG的合成和结构表征[78]Fig.5 (a) High-resolution XPS spectra of N 1s core level region of N-doped carbon[74];(b) synthesis and structural characterization of N-FLG[78]
在掺氮碳基催化剂的活性位的问题上,一些人认为存在于吡啶或吡咯的N位点[78].通过g-C3N4模板化策略对N结构和多孔结构的优化,Li等[79]开发了一种结构可调控的富氮多层石墨烯(N-FLG),阐明了吡咯-N的关键作用.如图5b所示,调节N-FLG的掺氮质量比后发现,OOH*和O*中间产物在C的K边缘光谱上的变化吸附曲线以及吡咯-N峰在N的K边氧烷光谱上负移.结果表明,该催化剂对电化学合成H2O2的选择性高达95%,吡咯烷-N含量与H2O2选择性呈正相关的线性关系.
而另一些人则认为石墨化的N位点是活性的中心[80-81].最近,Kim等[82]基于sp2碳位点为ORR活性位点的研究进一步揭示了P50碳纸支撑的掺氮氧化石墨烯(NrGO)的化学和结构性质;他们结合XPS、拉曼光谱和HRTEM等手段表征后发现,ORR催化活性中心在掺氮后仍为sp3碳区附近的sp2位点,这是由于N掺杂附近的邻碳在ORR环境中被H+或OH-等物质覆盖.此外,他们利用第一性原理理论对H2O2的机理进行了探究[76],发现轻度还原的氧化石墨烯无论在酸碱性溶液中对H2O2的选择性均达到了99%,但催化活性与溶液的pH值息息相关.另外他们还指出,发生耦合质子电子转移的决定性因素是费米能级的电子密度.
2.2.2 硫掺杂碳及其功能化
与纯碳材料相比,富硫电子的特性使掺杂硫的碳基复合材料有利于提高ORR中的电催化活性[83-86].先前的报告表明,在用硫掺杂碳基材料后,内部硫-碳界面处会产生电场,导致碳原子吸收更多的电子[87-88].这加速了催化剂中的电子传输,有效地促进了氧的吸附并进一步降低了过电势[89-91].
目前,引入硫的方法很多,包括原位包裹法[87]、硫的聚合物改性[92]、硫的物理化学吸附[87,93]等.Chen等[92]使用简单的两步二氧化硅模板方法制备了碳硫掺杂的空心多孔碳球-硫复合材料(HPCS-S).使用HRTEM分析,他们观察到球形碳上存在硫,平均颗粒尺寸为2~5 nm(图6a—f).他们还观察到高度结晶的硫纳米单晶.由于压缩应力,硫纳米晶体与碳球紧密结合,导致界面处的电子分布.在碱性溶液中,观察到硫化后多孔碳球的2e-ORR催化活性显著提高,并且H2O2的选择性从20%增加至72%.此外,硫掺杂将转移的电子数从3.5减少到2.7. H2O2的法拉第效率确定为70%,产率达到183.99 mmol·g-1. DFT计算表明,通过硫掺杂形成的C—S键本身表现出较低的过电势和较高的H2O2选择性.
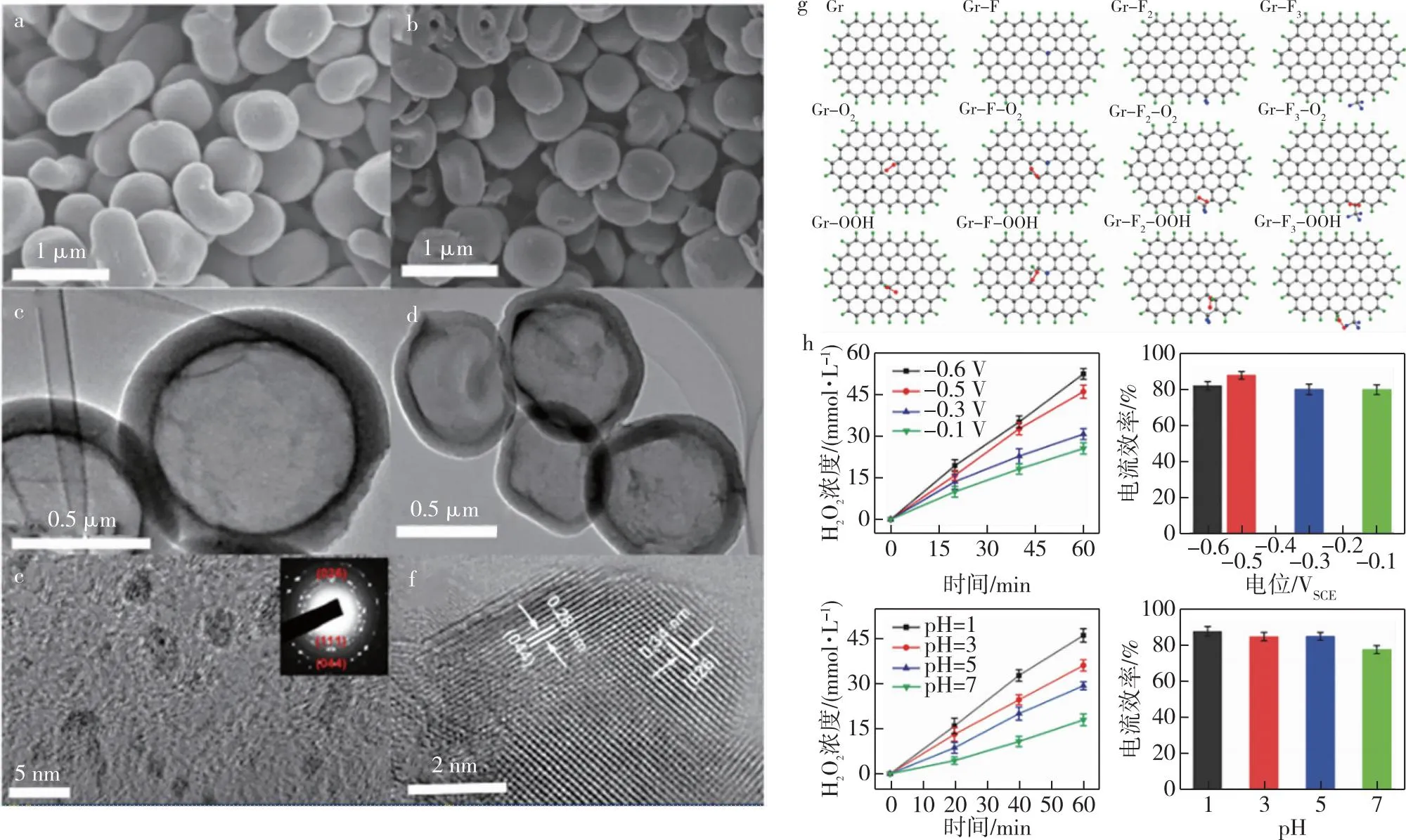
图6 (a—f) HPCS和HPCS-S的SEM图像、TEM图像和HR-TEM图像[92];(g) 不同类型的F掺杂和纯碳材料上O2和OOH吸附的计算模型[93];(h)NPC-1000上不同电位的H2O2浓度和电流效率[94]Fig.6 (a-f) SEM (up panels),TEM (middle panels),and HR-TEM (down panels) images of HPCS (left) and HPCS-S (right)[92];(g) calculation models of O2 and OOH adsorption on different types of F-doped and pure carbon materials[93];(h) concentration (left) and current efficiency (right) of H2O2 on NPC-1000 at different potentials (up panels) or different initial pH (down panels)[94]
用硫进行的表面改性可以改善碳基材料的电化学活性以及用于电解池的电极的润湿性[95-96].Perazzolo等[97]使用多种有机前体合成具有高表面氮的硫掺杂或共掺杂介孔碳(MCs).由于硫的电负性类似于碳的电负性,并且原子半径较大,因此特定浓度的硫掺杂可以提高MC的催化活性.但是,在高浓度(> 2%)下,增加的硫含量会影响O2排放的动力学,从而导致活性降低.值得注意的是,N-MC和S-MC在低pH值下表现出独特的催化活性.据报道,吡啶N的质子化/去质子化是增强N-MC的催化活性的关键,该机制影响ORR中间产物的吸附/解吸[98].
2.2.3 氟掺杂碳及其功能化
通常认为,H2O2的产率与催化剂的电子结构有关.由于电负性不同,上述氮、硫等杂原子掺杂碳材料中,具有较高电负性的氮原子可以通过破坏p共轭体系的完整性和诱导电荷再分配来激活碳p电子,从而改变碳材料的吸附特性[99].破坏电中性,引起电荷再分配,可以大大改善催化剂的电子结构,从而为氧还原生成过氧化氢创造更多的活性中心[72,77,100].在这种情况下,鉴于氟(F)的高电负性,可以诱导相邻的碳极化以产生活性中心并增强氧与碳之间的相互作用[90].同时,在碳材料中掺杂F可以诱导电荷极化,改变费米能级[101-102],调节电子转移性质,从而改变碳材料的吸附特性,有利于H2O2的生成[77].
先前的大多数报道认为,氟掺杂的碳纳米材料主要遵循氧的四电子途径[90].Zhao等[93]研究发现,H2O2选择性与OHH中间体在电催化表面的吸附能有关.他们报道了一种F掺杂多孔碳(FPC)催化剂,通过调节氟含量来影响碳材料的电子结构(图6g),即在碳骨架上形成共价结合的-CF2和-CF3物种.以这种方式调节O2分子活化和促进OOH*中间脱附,从而影响H2O2的选择性.
Pang等[99]在电Fenton降解有机污染物的应用研究再次表明,F的掺杂可以控制碳的电子结构,使电子转移途径趋于2e-.为进一步提高掺F碳基的催化活性,一些研究人员同时将两种活性位点引入碳骨架中,以提高电催化H2O2的性能.Chen等[103]设计了一种以聚偏二氟乙烯(PVDF)为模板,以多巴胺为氮源的N、F共掺杂空心纳米球(NF-Cs).理论模型计算表明,这种共掺杂结构被证实形成了C—F和C—N共价键.具有高电负性的F原子使介位的碳原子被活化,从而使其对OOH的吸附能力很高.N原子提高ORR催化活性和F原子提高ORR对H2O2选择性的结果表明,杂原子共掺杂的协同效应有助于实现碳基电催化剂对H2O2电催化的高效催化.
2.2.4 硼掺杂碳及其功能化
硼氮杂化体系可以提供独特的电子结构.在使用高硼和氮浓度的极端情况下,六方氮化硼(h-BN)在碳晶格内形成一些杂合的、随机分布的h-BN和C相畴,即硼碳氮混合体系(BCN)[104].
h-BN畴与石墨烯晶格界面的独特行为、表面在石墨烯衬底所具有的BCN构型与氧的结合能力较弱(ΔG<1.1 eV),导致OOH*中间体质子化,使H2O2成为反应主要产物[105].这种B和N浓度可调控体系,使得电催化过程优先通过2e-ORR来驱动生成H2O2.基于这一特性,这种B—C—N材料能够在电催化CO2还原、析氢反应和超级电容器电极等多个催化场景进行应用[106].
2.2.5 其他杂原子掺杂
类似于氮掺杂碳催化剂的催化机理,该催化剂激活π电子并引起电荷再分布,多元素掺杂碳、金属-氮共掺杂碳和碳化金属有机骨架(MOF)可以调节吸附/离解在碳材料上的O2和OOH*中间体,从而提高H2O2的选择性[107].
碳材料的多孔结构可以显著提高氧分子的传质速率并提供更多的活性位点,而金属有机骨架(MOF)是实现此目的的理想前体[95,107].Liu等[108]通过在2,6-二羧酸锌吡啶(ZnPDA)的吡啶前体上直接掺杂氮(图6h),并通过控制煅烧温度来制备具有可调节孔结构的N掺杂碳材料.多孔结构减少了中间体OOH-的停留时间并能够使H2O2从催化剂表面迅速解离,避免了H2O2进一步还原为H2O[109].
3 氧气还原生产H2O2的反应器开发
在迄今为止开发的众多有希望的电催化剂中,上述金属及非金属催化剂因其独特的反应特性而具有出色的H2O2选择性.但是,实验室条件与实际生产设备明显不同.此外,评估碳材料电催化性能的最常用方法是RDE/RRDE.RRDE电极的催化反应面积小,并且高速旋转有利于H2O2从催化剂表面的快速解吸,这不适用于商业应用(图7a).为了设计实际的电解槽以满足实际的生产要求,需要大的催化面积、高电流和高产量[110-112].
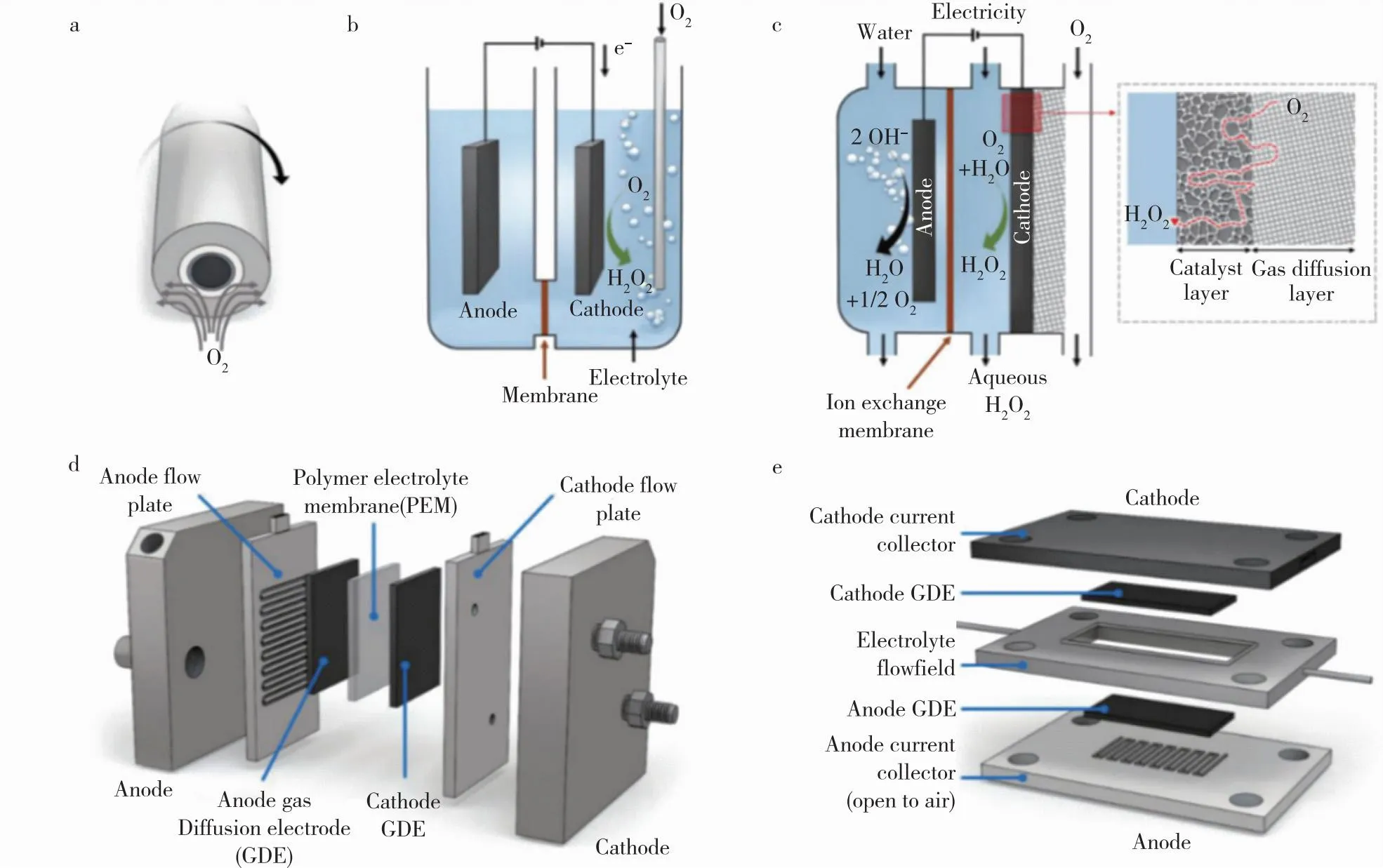
图7 电化学氧还原生产H2O2电解槽的设计进展[120]:(a) RRDE装置用于测定氧气还原H2O2的产率;(b) H-型电解池结构的示意图;(c) 连续流动池的示意图,其中催化剂沉积在GDE上;(d) 基于膜的流动反应器;(e) 微流体反应器Fig.7 Progress in the design of electrolytic cells for the production of H2O2 by electrochemical oxygen reduction[120]:(a) RRDE setup to measure yield of H2O2 from oxygen reduction;(b) schematic of an H-cell configuration;(c) schematic of a continuous flow cell with a catalyst deposited on a GDE;(d) membrane-based flow reactor;(e) microfluidic reactor
据报道,整个电解槽都浸入液体电解质中的H电解槽装置是生产H2O2的反应器(图7b).Yamanaka等[57]首次将质子交换膜(PEM)并入H电解槽装置中以产生H2O2.该PEM电解池可以在纯水中电合成H2O2,从而避免了电解液中其他离子的影响.但是,由于氧气在室温和常规压力条件下的溶解度较低(8 mg·L-1),因此两电子转移动力学受氧传质控制,从而限制了生产H2O2的催化反应的电流密度.气体扩散电极(GDE)可以将氧气直接供应到催化剂表面,而不受溶解氧浓度的限制,从而克服了与氧气传质速率有关的挑战[113].Li等[114]设计了一种PEM燃料电池,通过在Co-C复合材料上的GDE扩散,利用H2和O2连续产生H2O2,从而有效地净化了饮用水(图7c).
但是,在这种情况下,膜附近的H2O2局部浓度可能会很高.根据Yamanaka等[57]的方法,高的局部浓度进一步加速了H2O2的还原动力学或化学分解反应.Murayama等[115]通过暴露于固体聚合物电解质(SPE)直接合成H2O2,设计了一种由碳黑阴极催化剂、铂碳(Pt/C)阳极以及阴离子和阳离子交换膜(AEM和CEM)形成的“三明治”结构[116],具有磺酸基的苯乙烯-二乙烯基苯共聚物微球被用作固体电解液,并组装在阳极和阴极的两面上,以形成完整的电解槽进行测试(图7d)[117].在引入硝酸处理的炭黑纳米颗粒后,Perry等[5]开发了覆盖有10%表面氧的炭黑作为2e-ORR催化剂.具体来说,使用分别引入阳极和阴极负载在气体扩散层上(Sigracet 35 BC,Fuel Cell)、Nafion 115 薄膜(Fuel Cell Store)被夹在两个 PTFE 片之间以分隔腔室.在这项研究中包括功能化苯乙烯-二乙烯基苯共聚物微球或无机 CsxH3-xPW12O40.电化学产生的阳离子 (H+) 和阴离子 (HO2-) 在电场的驱动下穿过多孔电解质层并重新结合形成 H2O2.流经多孔电解质层的去离子水随后溶解不含杂质的 H2O2.观察到的结果表明,在0.438 V的起始电势下,H2O2的选择性增加到98%,优于迄今为止报道的最高的O2到H2O2转化率.根据文献报道,去离子水流速可以直接影响生成H2O2的浓度[115].根据实验分析,去离子水的流量固定为27 mL·h-1,可以直接获得20%的H2O2溶液.
此外,有报道指出,基于自然空气扩散的超疏水系统可提高氧气的传质速率(图7e),从而使反应器在空气中获得较高的H2O2产量成为可能[118].Zhang等[119]使用碳毡作为基材和扩散层,制备了自然空气扩散电极(NADE),使空气自然扩散到催化剂表面而无需外力.通过调节催化层的疏水性可以建立稳定的超疏水性三相界面.在不充气的情况下,因此获得了101.67 mg·h-1·cm-2的高H2O2产量.
4 结语
近年来,通过两电子氧还原途径生产H2O2已经成为一种环境友好且高效的电化学合成方法.新型的2e-ORR催化剂的理论模拟研究以及各类催化剂的相关结构设计已成为研究热点.本文综述了金属及非金属材料在H2O2生产中的最新应用(表1).通过对氧还原过程中间体的相对结合强度调节,以降低O—O键的断裂倾向,这种策略已被证明可以提高过氧化氢根等物种的选择性,并进而产生H2O2.进一步的研究证实,调节材料的局部原子结构,通过结合表面基团进行功能化以及掺杂外来原子也可以促进过氧化氢的产生[4,123].

表1 电合成H2O2电极催化剂的选择
通常认为ORR过程中的催化活性位以及中间体OOH*的电子转移机理起着至关重要的作用.研究人员已报道可以通过界面工程以及在表面上设计特定的官能团来增强OOH*的吸附能力并加速H2O2的解吸,从而优化现有的材料,进一步提高H2O2的选择性及活性.此外,本综述还总结了结构调整策略的研究,这些策略影响了掺杂后混合体的结构,例如C—S和C—N键的形成以及金属-氮-碳配位(M—N—C),可以改变碳原子的结构,将杂原子分散到独立的反应位点,有序的官能团可以有效地提高催化活性和H2O2的选择性[124].
尽管目前认为开发出很多先进材料在大规模生产H2O2时足以替代昂贵的贵金属,但仍需要克服许多挑战,包括结构调整、长期性能以及电极和反应器的实际设计.
1)金属氧化物和碳基复合材料仍需进一步发展.事实证明,负载在碳材料上的过渡金属氧化物具有出色的ORR活性(主要通过4e-途径)[125-126].这些材料包括钙钛矿型氧化物、尖晶石型氧化物和其他金属氧化物.然而,迄今为止,很少有金属氧化物与碳基材料偶联成为2e-ORR催化剂.由于改性材料保留了O—O键,因此可以预期金属氧化物复合材料的结构调整将迅速释放由具有高活性的金属氧化物产生的OOH*.
2)碳基材料的催化活性仍有很大的提高空间.如本文所述,碳基材料的表面改性可以产生许多活性位点,从而通过杂原子掺杂改善2e-ORR的催化性能.与碱性材料溶液中的Pt和Pd-Hg催化剂等商业材料相比,此类材料已显示出优异的ORR活性.但是,在中性电解质或弱酸性环境中,许多碳基材料通过两电子途径的氧反应无法与贵金属催化剂相比[127].
3)尚未完全理解碳基电催化剂的反应机理和活性中心.许多文献研究已经使用成熟的DFT技术探索了碳基催化剂的活性中心.但是,在确定H2O2生产过程中的活性位点时,这些结果仍然是模棱两可的.一方面,这可以归因于碳催化剂的复杂且非均质的结构.另一方面,由于不同的测试设备和表征环境,研究报告在相同的电解质中报告了不同的ORR活性,这导致对掺氮碳材料和相邻碳原子的活性中心的认识不明确.需要使用先进的高分辨率透射电子显微镜或原位拉曼技术进行进一步的电化学分析,以确定活性物质和氧还原反应的中心[128].
4)用于从碳基材料实际生产过氧化氢的电解池的设计需要进一步发展.迄今为止,已使用诸如RRDE、LSV、循环伏安法(CV)等实验室技术来评估大多数催化剂的电化学反应.但是,按比例缩放的RRDE电极的测试面积相对较小,并且需要特定的环境,因此不适合商业规模使用.为了满足工业生产的苛刻要求,有必要设计和建造适用于较大电流密度和输出功率的商业测试设备,例如膜流通池设备、燃料电池和固体电解质反应池.
总而言之,通过两电子ORR产生H2O2仍处于研究的早期阶段,具有机遇和挑战.特别是大多数 pH 依赖性、ORR活性和选择性研究都需要逐项筛选.虽然大多数碳基材料在碱性介质中可以表现出更高的双电子 ORR 选择性,但未来的研究工作应该集中在高活性金属与碳基材料偶联催化产生H2O2上,以提高整体反应活性.在设计高效催化剂、研究催化剂结构、活性位点之间的关系以及反应设备的实际应用等方面做出更多的努力,将进一步优化其催化性能,提高过氧化氢产率,并有助于实现大规模工业生产应用.
