WGCNA联合LASSO-COX方法筛选甲状腺癌预后关键基因及其临床价值分析
2023-12-11张澍漾郭松雪项承支飞虎谢立江赵萍
张澍漾,郭松雪,项承,支飞虎,谢立江,赵萍
1.绍兴市中医院 浙江中医药大学附属绍兴中医院普外科,浙江绍兴 312000;2.浙江大学医学院附属第二医院整形外科,浙江杭州 310009;3.浙江大学医学院附属第二医院甲状腺外科,浙江杭州 310009
甲状腺癌(thyroid cancer,THCA)发病率逐年增长,是全球增长速度最快的癌症之一,已经成为内分泌系统中最常见的恶性肿瘤。据统计,全球THCA患者已经超过5.86万例[1],女性的患病率是男性的3倍[2]。近30年,女性发病率上升了22倍,男性发病率上升了15倍,已经成为了威胁人民健康的十大癌症之一[3]。
近年来随着生物信息学的发展与应用,基因靶向治疗成为精准治疗肿瘤的热点。既往研究利用基因表达综合数据库(gene expression omnibus,GEO)和癌症基因组图谱(The Cancer Genome Atlas,TCGA)数据库确定了肺鳞癌的铁死亡差异表达基因(differentially expressed genes,DEGs),为癌症的治疗靶点和预后预测提供指导[4]。有研究通过免疫微环境、免疫治疗反应和基因突变分析等方法,构建了稳健的肾透明细胞癌症预后预测模型[5]。6-DNA甲基化特征是THCA患者无进展生存率的独立预后标志物[6]。Hsa-miR-139-5p是一种THCA预后标志物,参与HNRNPF介导的可变剪接,是一种与调节THCA主要信号通路和肿瘤毒性相关的新型调控机制[7]。GPX4表达的增加通过抑制铁死亡促进THCA的发生,且可以预测较差的临床结果[8]。SERINC2在乳头状THCA中可作为潜在的肿瘤驱动生物标志物[9]。目前我国已经常规开展了BRAF基因、TERT基因、RET/PTC重排和RAS基因等突变检测,用于THCA患者的辅助诊断[10]。
本研究通过TCGA-THCA数据集筛选出THCA和正常样本间的DEGs,随后进行权重基因共表达网络分析(weighted gene co-expression network analysis,WGCNA)和套索联系COX回归分析(least absolute shrinkage and selection operator regression COX analysis,LASSO-COX)获得5个与预后相关的关键基因,然后由关键基因构建预后预测模型,并采用免疫组化实验进行验证基因的表达,再进行受试者工作特征(receiver operating characteristic,ROC)曲线分析和生存分析,最后基于基因表达谱和风险评分进行基因集富集分析(gene set enrichment analysis,GSEA)用以评估相关途径和分子机制,为患者预后预测提供一个潜在选择。
1 资料与方法
1.1 数据收集与处理
在TCGA官方网站上收集2011年至2015年共511例THCA患者的临床数据和mRNA测序数据,共569份组织样本,其中THCA组织样本511份,甲状腺组织正常组织58份。随访时间至2015年,其中死亡人数为17例。使用Perl5.24.3软件将原始mRNA测序数据转换成mRNA表达矩阵。
1.2 差异基因的筛选
应用Limma R软件包对RNA-seq表达数据行归一化处理,并以P<0.01和|log2FC|>2为差异有统计学意义的阈值,进行差异基因表达分析筛选出THCA样本组和正常组之间的DEGs。
1.3 WGCNA
利用基因表达谱分别计算每个基因的绝对中位差(median absolute deviation,MAD),剔除了MAD最小的前50%的基因,利用R软件WGCNA包去除离群的基因和样本。首先,定义β(软阈值)的功效以确保标准的无标度网络,然后使用网络中每对节点基因与其Pearson相关系数之间的邻接值构造邻接矩阵,邻接矩阵用于计算拓扑重叠矩阵(topological overlap matrix,TOM)和相应的不相似度(1-TOM),基于TOM的差异度量,通过平均连接层次聚类构建树状图,将高度相似的模块聚集在一起,然后以0.25的高度截止合并。依据模块划分结果,计算模块与性状Pearson相关系数,选择与临床特性相关性最显著的模块。
1.4 LASSO-COX
结合患者的生存信息,首先对上述得到的关键模块基因进行LASSO-COX,减少基因之间共线性的影响,防止后续构建的风险模型变量过度拟合。LASSO回归分析通过引入惩罚系数(λ)将冗余变量的系数压缩为0,最后剩余的系数非零的变量为最终变量。本研究中使用5折交叉验证以确定最优惩罚系数,获取有效基因构建最佳预后模型。再将LASSO回归分析得到的基因进行多因素COX回归分析,计算每个多因素回归系数,构建风险评分方程。
1.5 免疫组织化学检测
2022年10月至12月在绍兴市中医院收集接受手术治疗的THCA患者的肿瘤组织和癌旁组织3例(女性2例,男性1例),进行免疫组化验证基因的表达。免疫组化采用常规两步染色法。组织通过石蜡包埋,然后切片脱蜡至水。微波加热修复抗原。滴加PBS洗净,滴加一抗。4℃孵育过夜,PBS冲洗干净,滴加二抗37℃孵育30min,DAB显色。苏木精再染色,脱水,透明,封片。所有药品均购自生工生物工程(上海)股份有限公司。
1.6 风险预后模型的建立与分析
根据上述LASSO-COX的结果,构建基于基因表达的风险评分方程。选择风险评分的最优截断值将患者分为高、低风险两组,进行Kaplan-Meier生存分析,利用R3.6.0软件绘制模型的ROC曲线,计算曲线下面积(area under curve,AUC),若AUC≥0.8,且P<0.05,则认为该模型具有较好的预测性能。利用R3.6.0软件绘制模型预测预后的列线图,以预测模型的C指数(C-index)评价模型的区分度。
1.7 根据模型评分分组进行GSEA分析
利用GSEA研究风险评分分组与京都基因和基因组百科全书(Kyotoencyclopdia of Genes and Genomes,KEGG)通路基因集的相关性。根据风险评分值的最优截断值将患者分成高、低风险两组,采用GSEA软件(version 3.0),设定参数:最小基因集为5,最大基因集为5000,1000次重抽样,以P< 0.05,FDR<0.25为差异有统计学意义条件,评估相关途径和分子机制。
2 结果
2.1 DEGs筛选
利用P<0.01和|log2FC|>2为筛选条件,在THCA组织-正常组间筛选到DEGs共计2130个,包含865个上调基因和1265个下调基因。
2.2 WGCNA分析
通过平均连通性图比较发现基因间联系软阈值为4后,设置模块合并阈值为0.25,绘制聚类树状图得到了9个关键模块。然后根据模块性状关系热图,发现青绿色模块与临床性状的关联性最高,确定其为关键模块,该模块包含403个基因。
2.3 LASSO-COX
整合生存时间、生存状态和基因表达数据,使用LASSO-COX对青绿色模块包含的403个基因进一步筛选,设置λ值为0.023,最终获得了5个关键基因LINC02550、STEAP2、ATP2C2、PLEKHG4B和SALL4,并构建模型公式。STEAP2的免疫组化结果显示该蛋白在肿瘤组织中的表达强于在正常组织中的表达(图1)。
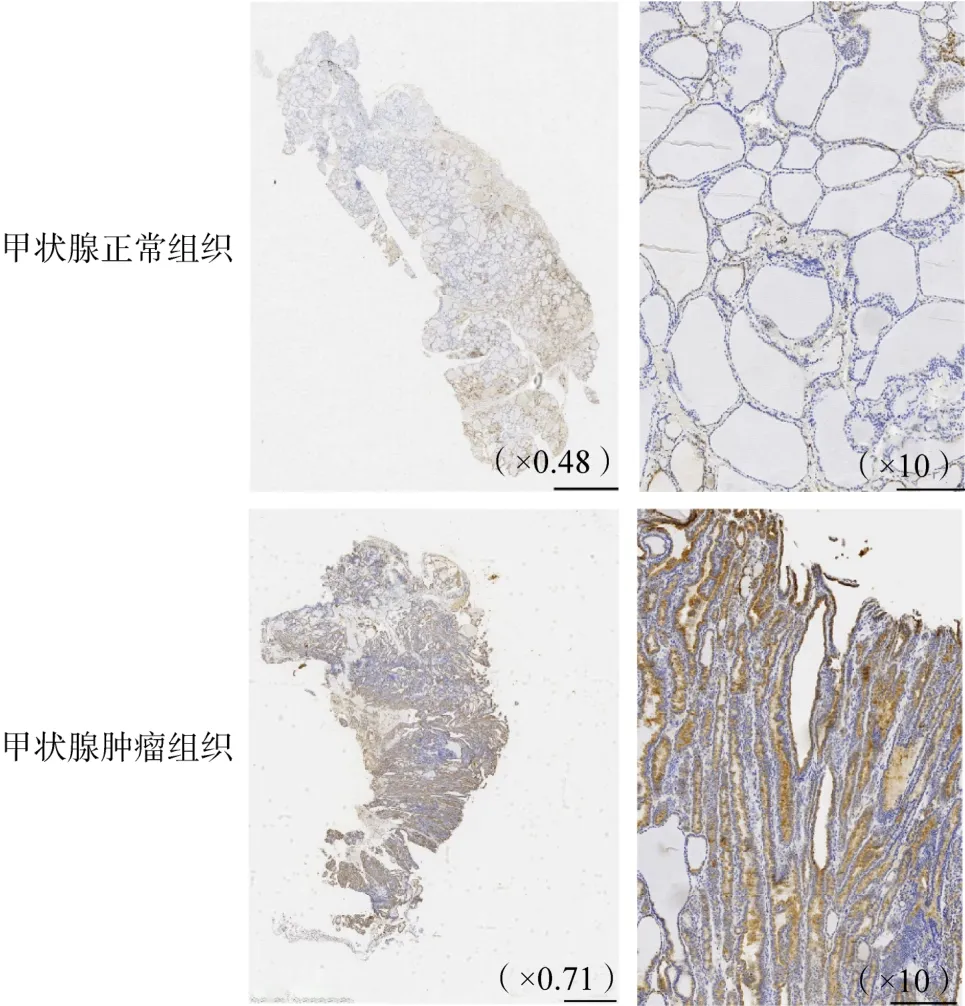
图1 STEAP2蛋白在甲状腺正常组织及肿瘤组织中的表达(DAB染色)
2.4 生存分析
生存分析表明,该风险评分显著影响THCA患者的预后,(P<0.001),并且高风险评分组的患者生存率较低(图2)。采用ROC分析获得1、3、5年曲线下面积分别为0.93、0.88、0.86,均大于0.8,说明该模型具有良好的预测性能(图3)。
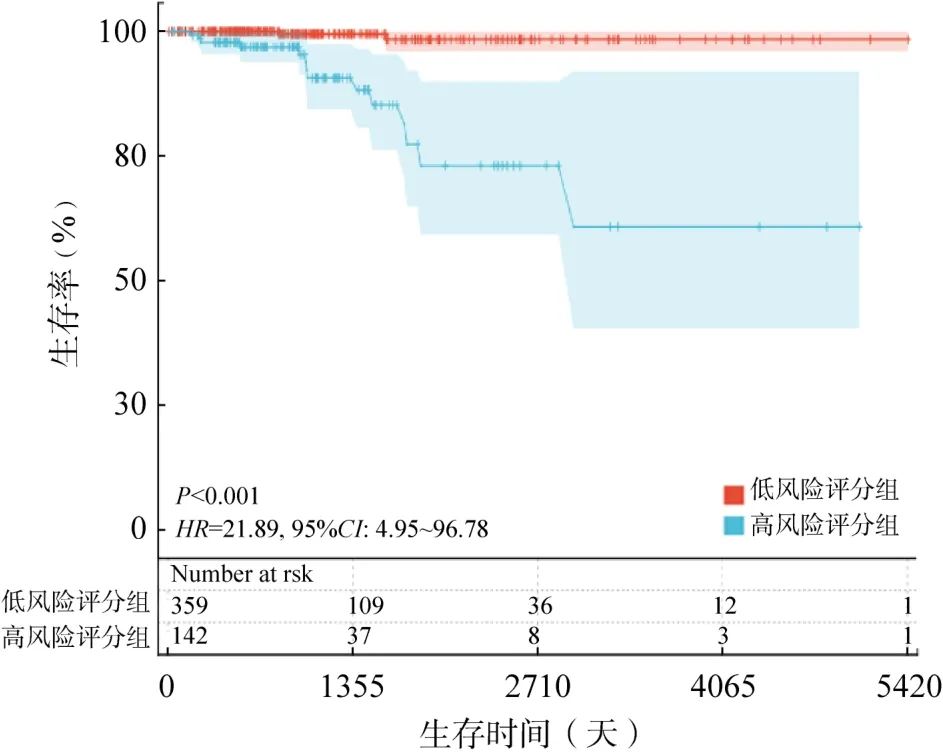
图2 预后基因风险评分模型的生存分析
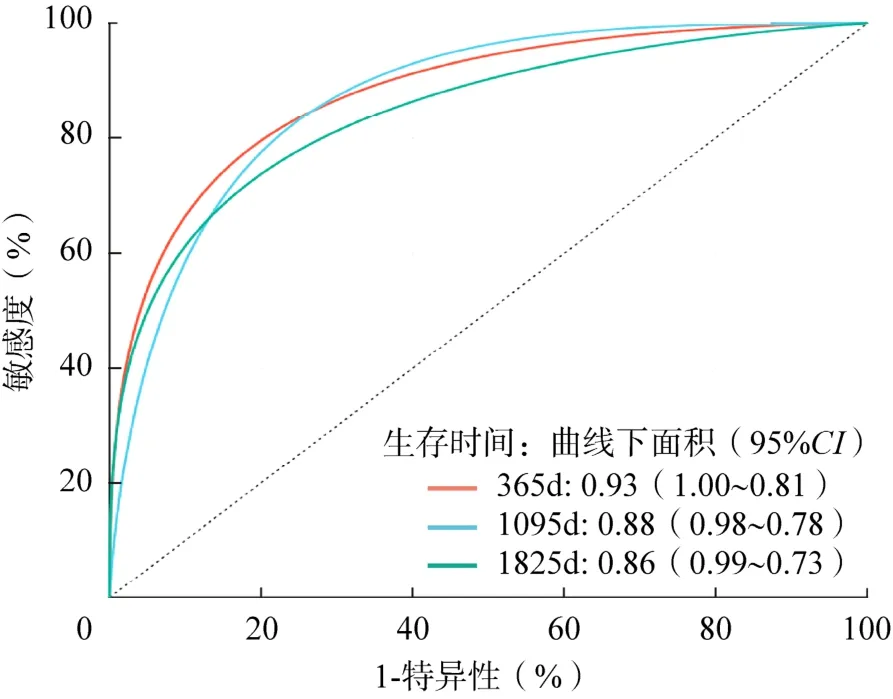
图3 预后基因风险评分模型的ROC曲线
2.5 诺莫图
基于以上结果利用R3.6.0软件绘制基于5个基因的综合风险评分和主要临床特征的组合预测THCA患者生存的列线图,结果显示列线图对THCA患者具有良好的预后预测能力(C-index=0.96,图4)。
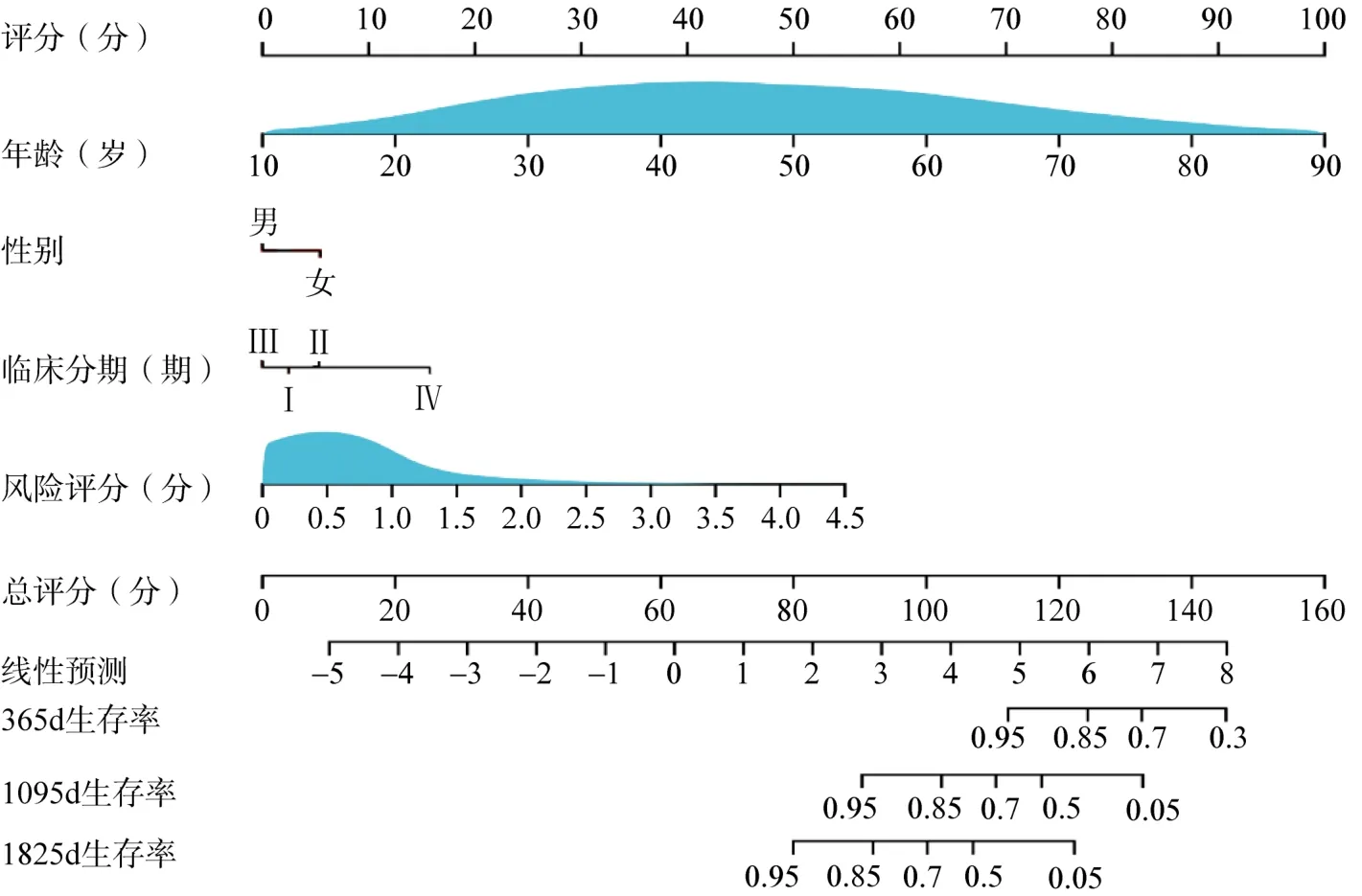
图4 风险评分联合临床特性建立的预测患者生存诺莫图
2.6 GSEA分析
5基因构建的预后预测模型具有良好的预测性能,因此认为该模型可能与影响预后的信号通路有关。GSEA分析获得关键通路mTOR信号通路、Hedgehog信号通路、细胞自噬调节及转化生长因子-β信号通路,且均富集在高风险评分组(图5)。
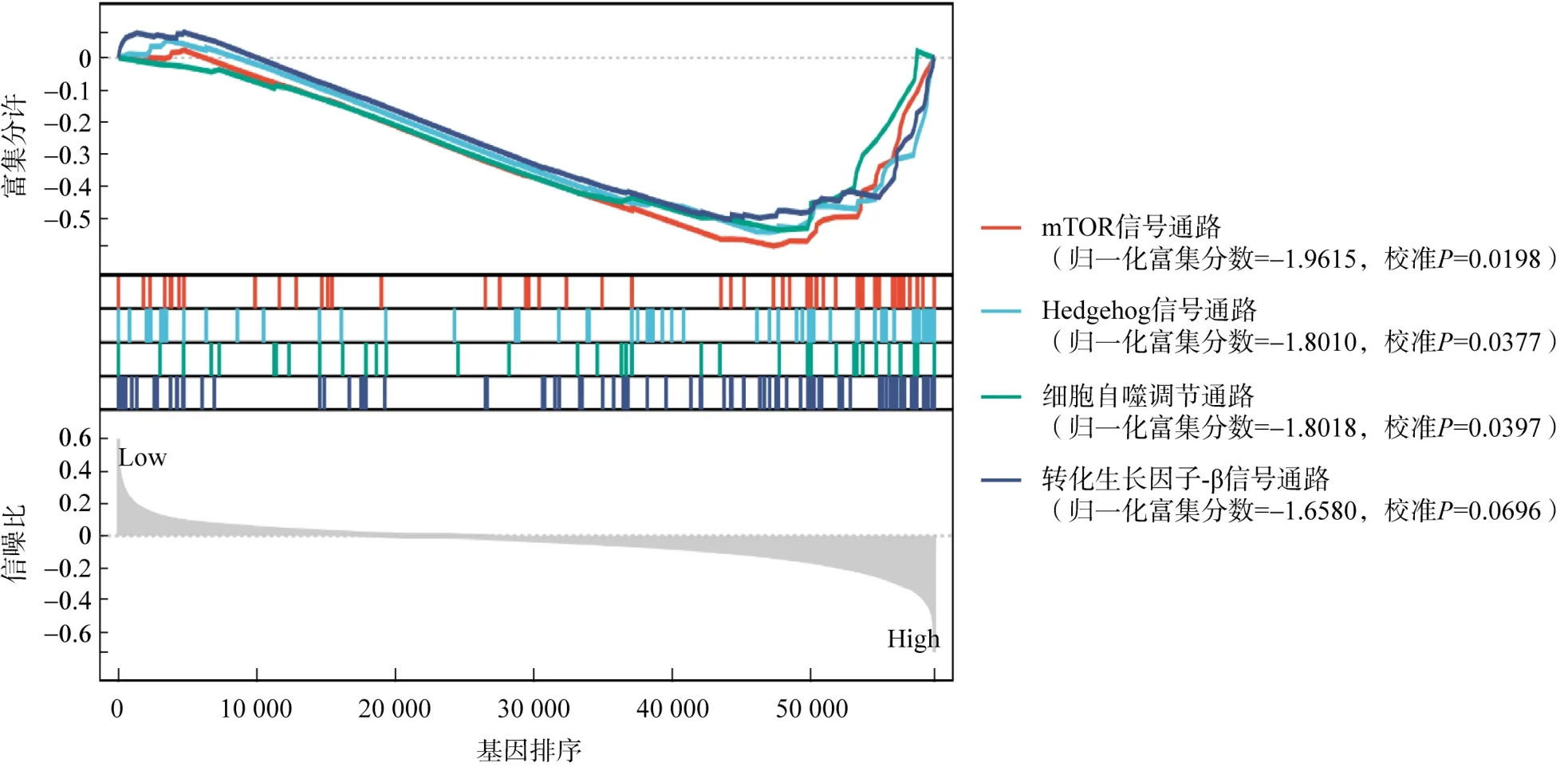
图5 GSEA富集信号通路
3 讨论
THCA在内分泌恶性肿瘤患者中占比超过90%,且女性患病率显著高于男性,因为雌性激素有可能是甲状腺良恶性细胞的一种有效生长因子[11-12]。基因突变是THCA发生、发展的重要因素之一,并与其疾病诊断、预后评估及分子靶向药物治疗密切相关。早期对THCA进行风险等级评定,不仅避免了对轻度患者过度治疗,而且有利于精准预测患者的预后[13]。
本研究结果表明,LINC02550、STEAP2、ATP2C2、PLEKHG4B和SALL4这5个基因与THCA预后紧密相关。关于LINC02550的研究目前还没有报道。STEAP2作为STEAP家族的成员,是重要的体内金属还原酶,在维持铁稳态中发挥重要作用,对不同的癌种有不同的响应[14]。本研究中该基因在甲状腺肿瘤组织中是下调的,免疫组化结果显示该蛋白在甲状腺肿瘤组织中高表达,与已报道研究发现其在乳头状THCA中的低表达相反,可能由于该蛋白在甲状腺不同类型细胞的表达不同[15]。
ATP2C2基因与许多重要的生物学功能有关,如调节钙稳态、调节语言障碍中的语音短期记忆[16-17]。既往研究发现ATP2C2可能是乳腺癌患者肿瘤微环境状态的指标[18-19]。PLEKHG4B在细胞之间连接成熟期间参与肌动蛋白细胞骨架重塑,在甲状腺肿瘤中PLEKHG4B表达是下调的,可能由于其下调,增加了甲状腺肿瘤细胞间的黏附力形成时间,有益于其转移[20]。SALL4是公认的致癌基因,该基因不仅通过Wnt/β-catenin和PTEN/PI3K/AKT通路调节细胞生长,还通过细胞凋亡相关的基因,如CYC3和CUL3的表达影响细胞凋亡[21-22]。SALL4在不同的癌种中起不同的作用,在乳腺癌和肝癌中,通过激活Wnt/β-catenin信号,促进癌细胞的增殖、迁移和侵袭;在胃癌中激活转化生长因子-β/SMAD信号通路,促进癌细胞转移[23-25]。研究表明,SALL4基因在THCA组织中表达上调,可能参与肿瘤细胞的发展和转移[26]。
GSEA分析表明mTOR信号通路、Hedgehog信号通路、细胞自噬调节、转化生长因子-β信号通路富集在高风险评分组。Hedgehog信号通路作用于哺乳类动物的胚胎发育和组织分化,控制细胞的生长与增殖。该信号通路异常激活与多种恶性肿瘤的发生有密切关系,参与肿瘤增殖、迁移、血管生成、上皮-间质转化、肿瘤干细胞调控等,有研究表明在THCA患者中通过抑制Hedgehog信号通路可以激活TGF-β信号通路,从而抑制肿瘤细胞的凋亡[21]。此外,TGF-β是上皮-间质转化过程的主要开关,可通过很多信号通路来诱导上皮-间质转化的发生。
综上所述,本研究通过生物信息学方法分析THCA基因表达谱数据,筛选了5个关键基因可为潜在预后标志物,并通过免疫组化实验进行表达验证。此外,建立了生存预后模型,有助于THCA的预后生存预测与基因靶向治疗。
