碘催化下氯苯的氯代反应
——用理论计算化学方法深度分析一道高考试题
2023-12-11柏一慧毛倩芸张恬王瑞哲
柏一慧,毛倩芸,张恬,王瑞哲
1 浙江师范大学化学与材料科学学院,浙江 金华 321004
2 浙江师范大学教育学院,浙江 金华 321004
本文探讨的问题来自于一道高考试题,2021年全国统考高考化学试卷(浙江卷) (1月选考),第29题第(5)小问;原试题如下[1]:
在一定温度下,以I2为催化剂,氯苯和Cl2在CS2中发生平行反应,分别生成邻二氯苯和对二氯苯,两产物浓度之比与反应时间无关。反应物起始浓度为0.5 mol·L-1,反应30 min测得氯苯15%转化为邻二氯苯,25%转化为对二氯苯。保持其他条件不变,若要提高产物中邻二氯苯的比例,可采用的措施是____。
A.适当提高反应温度 B. 改变催化剂 C. 适当降低反应温度 D. 改变反应物浓度
问题解答:
根据k1/k2= (0.5 × 15%)/(0.5 × 25%) = 3/5
由此可以推出生成邻二氯苯的反应速率常数小于生成对二氯苯的反应速率常数,其对应的活化能更大。由于升高反应温度更有利于活化能大的反应,因此提高反应温度可以提高产物中的邻二氯苯的比例;另外如果改变催化剂,使生成邻二氯苯的活化能降低得更多,也可以提高邻二氯苯的比例。因此,A、B正确。考点:化学反应速率的影响因素。
而该反应体系的真实情况到底是怎样呢?在碘催化下,升高温度真的能提高邻二氯苯的比例吗?碘在此处到底是如何起到催化作用的?反应过程中的能量变化是怎样的?带着这些问题,我们对深入了解该反应体系产生了兴趣。经查阅文献,我们发现H. F. Wiegandt在“Improved Yields ofp-Dichlorobenzene. Substitutive Chlorination of Benzene”[2]一文中指出,碘催化氯苯二氯代时与其他几种催化剂(如AlCl3、FeCl3、SbCl5等)的催化情况有所不同,在较高温度下,对二氯苯的比例会增加,而邻二氯苯的比例则会下降;而其他几种催化剂作用下,情况则是相反的。如果以该文献中的实验结果作为本文前面提到的高考题的参考数据,则该试题存在违背实验事实的现象,需要做修正,否则不宜再出现在高考试题中。
由于H. F. Wiegandt的实验结果是基于苯的氯代反应体系,起始反应物为苯,反应体系中缺少试题中提到的溶剂CS2,因此两者的反应环境有所不同。那么溶剂CS2的加入是否会导致该反应体系发生一些变化,从而导致更高温度下邻二氯苯的比例更高?为了进一步验证该试题的合理性,也为了使学生在教学过程中更加深入了解苯的取代反应机理,本文将借助理论计算化学的方法,对碘催化下氯苯发生氯代反应的反应机理进行探讨,对反应过程中各物质结构和能量的变化进行模拟。
1 计算模型和计算方法
使用量子化学密度泛函理论(DFT)[3]的B3LYP方法[4]对反应体系中的反应物、生成物、中间体及过渡态的几何构型进行全优化,对碘采用赝式基组(LANL2DZ)[5],对其他原子采用6-311g(d,p)基组[6];同时在同一理论水平下对势能面上的全部驻点进行了振动频率分析,以确认它们为势能面上的稳定驻点。为了证明每个过渡态的正确性及势能面上连接着两个所期望的能量最低点;通过内禀反应坐标(IRC)分析,进一步验证了过渡态的可靠性并确认了反应路径。此外,为了获得更加准确的能量值,在B2PLYPD3/def2TZVP水平下计算各物种的单点能(I使用LANL2DZ基组),并对总能量进行了零点能校正[7]。溶剂化效应采用基于密度(SMD)的溶剂化模型[8]。所有的计算都用Gaussian 09程序包[9]来完成,并采用GaussView 5.0.8作为可视界面。
2 结果与讨论
2.1 碘催化下氯苯二氯代的反应机理
对于该反应体系,首先需要解决的问题是:碘作为催化剂,其真实作用是什么?真正起催化作用的到底是不是碘单质?碘作为催化剂与其他路易斯酸催化剂有什么不同?
在氯苯的氯代反应中,首先需要一个带正电荷的氯原子,该氯原子应该来自于极化的氯气分子,而体系中催化剂的作用主要就是提供该带正电荷的氯,因此碘的作用是对氯分子进行极化并以此提供带正电的氯,起催化作用的碘需要缺电子,起到类似于路易斯酸的作用。因此,该反应体系中起催化作用的碘可能以ICln的形式存在,n可以为1,3,5等。为确定真正起催化作用的碘代物,对各种碘分子、氯气分子、碘氯复合物、各种氯代碘化合物的结构进行了优化,对它们的相对能量进行了比较,相关结果在表1中列出。
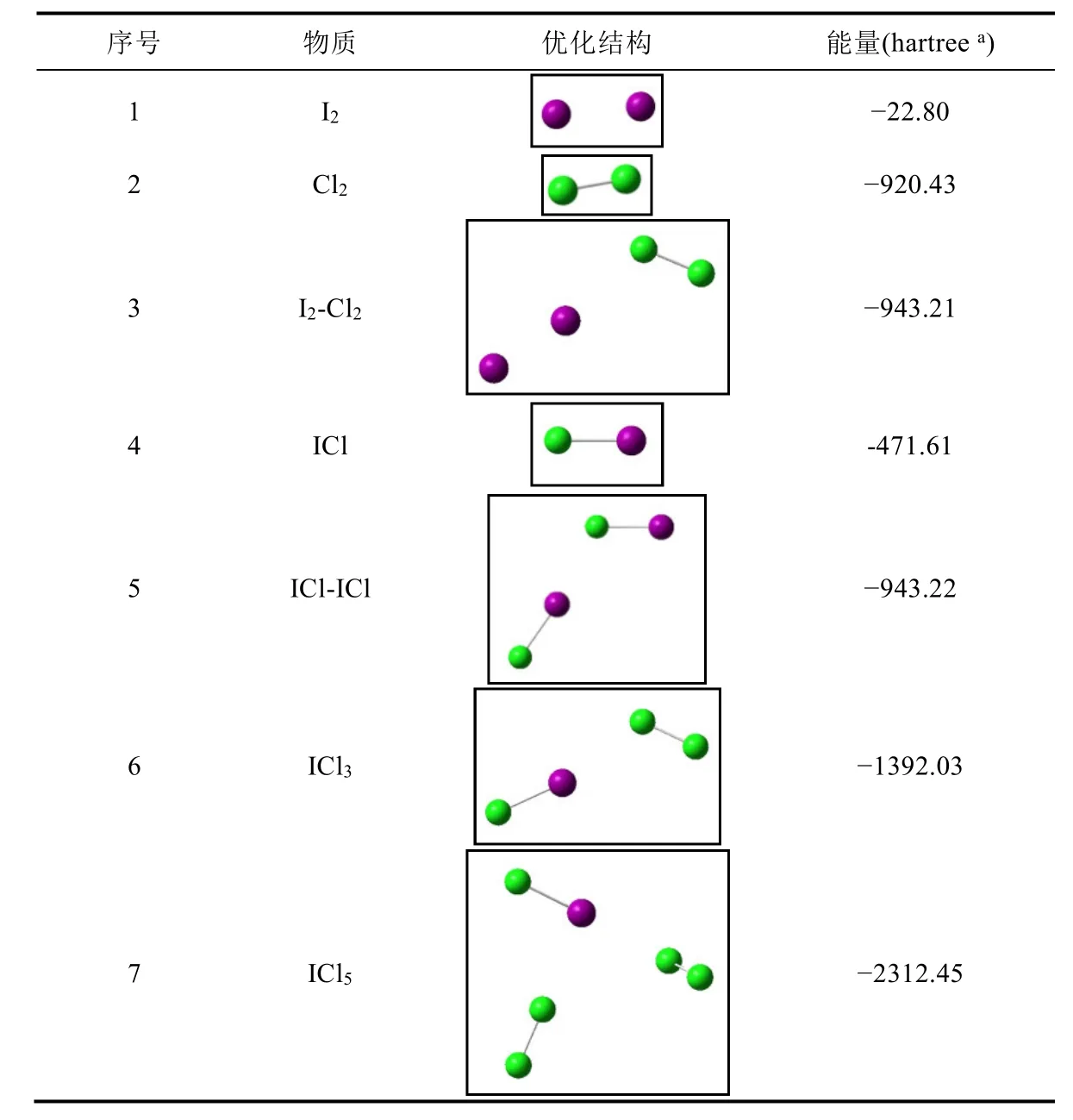
表1 碘、氯及氯代碘的优化分子结构与能量
从表1的结果可以分析得出,2分子ICl的复合物(ICl-ICl)比碘氯复合物(I2-Cl2)低8.2 kcal·mol-1(1 kcal·mol-1= 4.2 kJ·mol-1),说明I2与Cl2反应生成ICl在能量上是有利的;ICl3优化后的结构显示是ICl和Cl2的复合物,其能量比孤立的ICl和Cl2的能量之和高5.5 kcal·mol-1,说明ICl3不稳定,难以形成三个牢固的I—Cl键;ICl5优化后的结构显示是ICl和2分子Cl2的复合物,其能量比孤立的ICl和2分子Cl2的能量之和高11.1 kcal·mol-1,说明ICl5也不稳定,不存在五个牢固的I—Cl键。
因此,推测碘在本反应中起催化作用的主要是ICl。此外,John T. Stock在“Arthur Slator and the Chlorination of Benzene”一文中也提到,在苯-氯气-碘的体系中ICl是反应的催化剂[10]。结合表1中计算得到的氯气-碘体系中各种可能物种的相对能量分析和该文献中给出的信息,在本文的反应机理研究中,采用ICl作为催化剂。
2.2 氯苯在碘催化下氯代的反应机理
在ICl催化作用下,氯苯可以发生芳香亲电取代反应,其反应机理和主要反应路径如图1所示。首先,碘与氯气作用生成ICl,ICl与氯气作用,使氯气分子发生极化,其中一个氯原子带部分正电荷,该正性的氯接下来作为亲电试剂进攻氯苯的苯环,根据定位规则,氯苯的邻、对位电子云密度较高,接受该正电性氯的进攻,分别经过两个过渡态o-TS1和p-TS1,生成中间体o-IM1和p-IM1;σ-络合物o-IM1和p-IM1脱去氢质子经过过渡态o-TS2和p-TS2最终生成o-Pro和p-Pro。

图1 氯苯在一氯代碘催化下发生二氯代的反应机理图
首先研究了气相下该反应体系的反应机理,对该反应机理中两种反应路径所涉及到的物质和过渡态进行了优化,各物质的优化结构图见图2。

图2 氯苯在一氯代碘催化下发生二氯代的物质优化图(气相反应)
从图2中给出的优化结构可以看到,ICl首先与氯气作用,使氯气分子发生极化,Cl—Cl键被削弱,其中一个氯原子靠近碘原子,另一个氯原子接近氯苯的邻位或对位碳,形成反应复合物(o-Re/p-Re),接下来该氯原子与氯苯的邻、对位碳进一步靠近,被进攻的碳原子离开原来的分子平面,产生过渡态(o-TS1/p-TS1),可以看出,邻位取代的过渡态o-TS1中与苯环上的两个氯在空间上相对于对位取代的过渡态p-TS1更分散,p-TS1中靠近氯苯的C1原子,使C1也离开原来的苯环平面,共轭体系进一步遭到破环,因而p-TS1相对o-TS1能量更高;接着进攻的氯原子与氯苯的邻、对位碳结合生成σ-络合物(o-IM1/p-IM1);中间体o-IM1和p-IM1中的进一步靠近sp3杂化的C原子,变成更稳定的o-IM2和p-IM2,接着攫取sp3杂化的C原子上的氢质子,经过过渡态(o-TS2/p-TS2);最后生成产物复合物(o-Pro/p-Pro),产物复合物为邻二氯苯或对二氯苯与ICl、HCl的混合物。
对各反应物质的能量进行了分析,并制作了反应能量变化图,如图3所示。可以看出反应分两步,第一步是中间体σ-络合物的生成,第二步是σ-络合物脱质子生成产物。从图3可以得出,生成σ-络合物的过渡态(o-TS1、p-TS1)对应的活化能分别为26.4、31.2 kcal·mol-1,p-TS1相对o-TS1能量更高,邻位氯代这一步的活化能比对位氯代的低4.8 kcal·mol-1;σ-络合物脱质子生成产物复合物的过渡态(o-TS2、p-TS2)对应的活化能分别为14.1、1.7 kcal·mol-1,邻位氯代这一步的活化能比对位氯代的高12.4 kcal·mol-1,这一步对位氯代的路径更容易;产物复合物o-Pro的相对能量比p-Pro高2.3 kcal·mol-1,对位取代的产物更稳定。

图3 氯苯在一氯代碘催化下发生二氯代的反应能量图(气相反应)
可以看出,在氯苯发生氯代的两步反应中,第一步的活化能远大于第二步的活化能,为反应的决速步骤。在决速步骤中,氯苯邻位氯代的活化能明显低于对位氯代的活化能,说明氯苯邻氯代比较容易,在这种情况下,如果要提高邻二氯苯的比例,应该控制反应温度相对比较低更好。
气相下的反应能量变化是如此,那么如果考虑溶剂化作用,氯苯邻位氯代和对位氯代的反应能量变化是否会与气相下有较大的不同?为此,接下来对溶剂条件下的反应能量变化进行了研究。
反应如在液相中进行,通常需要考虑溶剂化效应,本文中溶液中溶质的自由能Gsoln由以下公式来得到[11]:
式(1)中Ggas为气相状态下物质的自由能;ΔGsolv为溶解自由能,在SMD模型下采用M05-2X/6-31G*方法得到[12];ΔG’表示在298.15 K时将1个标准大气压的1 mol理想气体溶解成1 mol·L-1液体时自由能的变化,为1.89 kcal·mol-1。
因为本文中提到的高考试题中的反应是以CS2做溶剂,因此进一步研究了以CS2为溶剂时该反应体系的反应机理,该反应机理中两种主要反应路径所涉及到的物质和过渡态的优化结构、各反应物质的相对能量以及物质的能量变化如图4所示。

图4 氯苯在一氯代碘催化下发生二氯代的反应机理图(以CS2为溶剂)
在以CS2为溶剂的反应体系中,第一步生成邻位、对位取代的σ-络合物的活化能分别为30.5、34.4 kcal·mol-1,邻位氯代这一步的活化能比对位氯代的低3.9 kcal·mol-1;第二步脱质子的步骤中,邻位、对位取代的活化能分别为19.3、2.5 kcal·mol-1;邻位取代产物的相对能量比对位取代产物高2.3 kcal·mol-1。在该溶液体系中,在决速步骤中,氯苯邻位氯代的活化能依然低于对位氯代的活化能,因此,在这种情况下,如果要提高邻氯二苯的比例,依旧应该控制反应温度相对比较低更好。
从以上关于氯苯在碘催化下发生氯代的反应机理的理论研究我们可以看出,无论是在气相条件下还是在以CS2为溶剂的反应体系中,氯苯发生对位氯代的活化能都要大于邻位氯代的活化能,从而可以推出在较高温度下,对二氯苯的比例会增加,这与Herbert F. Wiegandt在文献中给出的实验事实一致,而这个结果与本文前面提到的高考化学试题的答案却是相反的。因此可以给出的建议是,该高考试题有必要做进一步的修正,以确保其科学性。
3 结语
本文采用理论计算化学方法,对碘催化下氯苯的氯代反应的反应机理进行了研究,对涉及该反应体系的一道高考试题进行了深入的分析,得到了如下结论:
(1) 碘催化下氯苯的氯代反应中主要起催化作用的应该是ICl。
(2) 该反应体系在气相条件下,在其决速步骤中,氯苯邻位氯代的活化能明显低于对位氯代的活化能,说明氯苯邻氯代比较容易;如果要提高邻氯二苯的比例,应该控制反应温度相对比较低更好。该理论研究结果与文献提供的实验结果一致。
(3) 该反应体系在以CS2为溶剂的反应条件下的结果与气相条件下的结果保持一致。
(4) 该相关高考试题建议可做进一步的修正,以确保其科学性。
