波齐替尼合成工艺研究
2023-12-06焦小雨唐春雷
袁 昕 焦小雨 唐春雷
(江南大学 生命科学与健康工程学院,江苏 无锡 214122)
波尼替尼(Poziotinib,1),化学名1-(4-(4-(3,4-二氯-2-氟苯胺)-7-甲氧基喹唑啉-6-氧基)-1-哌啶基)丙-2-烯-1-酮(图1),是由韩国首尔医科大学于2008年研发的一种泛人表皮生长因子受体2(EGFR2)酪氨酸激酶抑制剂[1],主要靶向EGFR野生型、EGFRT790M/L858R、HER2和HER4,对应的IC50分别为3.2、2.2、5.3和23.5 nmol·L-1[2]。据数据显示,目前非小细胞肺癌(NSCLC)患者中90%的HER2突变是20号外显子突变,4%的患者中存在EGFR和HER2的20号外显子双突变[3]。然而由于20号外显子的突变改变了药物结合口袋的大小、限制了大的刚性抑制剂的结合,目前已有的许多酪氨酸激酶抑制剂,如阿法替尼[4]、达克米替尼、来那替尼[5]等,对EGFR和HER2的20号外显子突变抑制效果不佳。因此针对EGFR和HER2的20号外显子双突变的NSCLC患者依旧缺乏有效的治疗方法。而Poziotinib由于结构较小、存在可灵活旋转结构的特点,可以规避空间位阻变化[3],给患者带来良好的治疗效果。根据最新的Ⅱ临床试验数据(NCT03066206),Poziotinib在EGFR和HER2的20号外显子突变的NSCLC中获得了30%的客观缓解率[6],有望成为具有良好治疗效果的酪氨酸激酶抑制剂。作者对Poziotinib的合成方法进行了研究,以期为该化合物的合成以及结构类似物的研究提供理论参考。
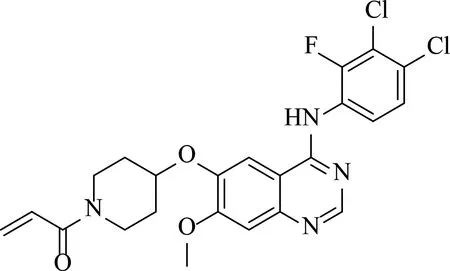
1图1 Poziotinib的结构Fig. 1 The structure of Poziotinib
1 合成路线
目前原研公司报道的Poziotinib(1)的合成路线如图2所示[7,8],以2-氨基-4,5-二甲氧基苯甲酸(2)为原料在210 ℃下与盐酸甲脒反应得到6,7-二甲氧基喹唑啉-4(3H)-酮(3)。化合物3在甲磺酸中与甲硫氨酸反应得到6-羟基-7-甲氧基喹唑啉-4(3H)-酮(4)。化合物4在吡啶和无水乙酸中乙酰化生成关键中间体3,4-二氢-7-甲氧基-4-氧代喹唑啉-6-醇乙酸酯(5)。化合物5与三氯氧磷在甲苯中经氯化得到4-氯-7-甲氧基喹唑啉-6-乙酸酯(6)。化合物6在含氨水的甲醇溶液中去乙酰化得到4-氯-7-甲氧基喹唑啉-6-醇(7)。化合物7与4-(甲苯-4-磺酰氧)哌啶-1-羧酸叔丁酯(11)经Mitsunobu反应得到4-((4-氯-7-甲氧基喹唑啉-6-基)氧基)哌啶-1-羧酸叔丁酯(8)。化合物8在乙腈溶液中与3,4-二氯-2-氟苯胺(12)经取代反应得到4-((4-((3,4-二氯-2-氟苯基)氨基)-7-甲氧基喹唑啉-6-基)氧基)哌啶-1-羧酸叔丁酯(9)。化合物9以丙酮作为溶剂通过盐酸脱去叔丁基羰基(Boc)的保护得到N-(3,4-二氯-2-氟苯基)-7-甲氧基-6-(哌啶-4-氧基)喹唑啉-4-胺(10)。化合物10在碳酸氢钠的四氢呋喃和水的混合溶液中与丙烯酰氯经酰化反应得到Poziotinib (1),总收率12.7%(以2计,以5计收率为32.0%)。该路线操作复杂、反应耗时长、副产物多、成本较高,不利于工业化生产。

图2 化合物1的原研合成路线Fig. 2 Original synthetic route of compound 1 in Literature
原研专利合成路线共有9步,经成本核算,其中经1~3步得到化合物5所需成本高于直接商业化获得化合物5的价格。化合物2至化合物4的制备步骤危险性较大。这几步反应均在无溶剂且高温的情况下进行,反应可能不均匀,且氯代后的产物6往往结构不稳定,暴露在空气中容易与空气中的水发生取代反应变回氯取代前的原料。故改进工艺路线(如图3所示)中6优先与3,4-二氯-2-氟苯胺(12)进行亲核取代反应,有效解决了化合物稳定性低的问题,相比原研路线,收率也得到了保证。改进后的工艺路线以廉价易得的7-甲氧基-4-氧代喹唑啉-6-醇乙酸酯(5)为起始原料,经6步反应得到Poziotinb,且收率由原来的12.7%提高至42.3%(以5计)。另外,改进路线在关键中间体9和Poziotinib(1)粗品的纯化上采用重结晶的方式,节约了时间和溶剂成本。优化后的工艺路线在提高总收率的前提下,降低了生产成本和能耗,反应条件相对温和,后处理操作简便,适合工业化大量制备。
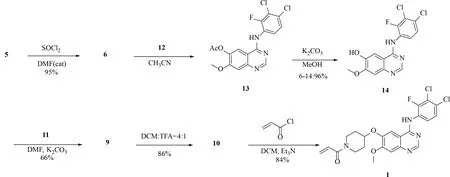
图3 化合物1的改进合成路线Fig. 3 Improved synthetic route of compound 1
2 实验部分
2.1 主要仪器与试剂
7-甲氧基-4-氧代喹唑啉-6-醇乙酸酯(5),97%;4-(甲苯-4-磺酰氧)哌啶-1-羧酸叔丁酯(11),98%;3,4-二氯-2-氟苯胺(12),98%。以上试剂均购于上海毕得医药有限公司。实验所用试剂均为市售分析纯或化学纯,未经进一步纯化处理。TLC展开剂和溶剂比例均为体积比。
液相-质谱仪,LC-MS-80,Waters公司,ESI离子源,正离子MS模式,离子源温度120 ℃,锥孔电压40 V,碰撞能量20 eV,全扫描质荷比为50~1 500 Da,扫描时间为0.5 s。液相色谱仪,LC1260,安捷伦科技有限公司。核磁共振仪,Bruker AVII-400 MHz,德国Bruker公司,TMS为内标。
2.2 合成步骤
2.2.1 4-氯-7-甲氧基喹唑啉-6-乙酸酯(6)
将7-甲氧基-4-氧代喹唑啉-6-醇乙酸酯(5)(30.0 g,128.1 mmol)和氯化亚砜(300 mL)置于1 000 mL反应瓶中,随后加入催化量的N,N-二甲基甲酰胺(DMF,0.15 g),80 ℃搅拌反应2 h,TLC检测反应(展开剂:石油醚-乙酸乙酯,体积比1∶1),减压蒸除溶剂后,冰浴下向残留物中缓慢滴加饱和碳酸氢钠水溶液,调pH值至8~9,用二氯甲烷(3×500 mL)萃取,合并有机相,有机相水洗(3×300 mL),饱和氯化钠溶液(3×300 mL)洗涤,无水硫酸钠干燥,抽滤,滤液减压浓缩得到得灰色固体粉末(6)(30.6 g,收率95%)。ESI-HRMS(m/z): 253.05 [M+H]+。1H NMR (400 MHz, CDCl3)δ: 8.97 (s, 1H), 7.92 (s, 1H), 7.46 (s, 1H), 4.05 (s, 3H), 2.42 (s, 3H)。
2.2.2 4-((3,4-二氯-2-氟苯基)氨基)-7-甲氧基喹唑啉-6-羧酸酯(13)
将中间体6(30.0 g,75.9 mmol)和乙腈(300 mL)置于1 000 mL反应瓶中,再加入(14.9 g,83.4 mmol)的3,4-二氯-2-氟苯胺(12),室温下搅拌4 h。TLC检测反应完全(展开剂:二氯甲烷-甲醇,体积比20∶1),反应结束后,有固体析出。抽滤,用乙腈(60 mL)洗涤滤饼。滤饼经真空干燥(45 ℃,24 h)后得白色粉末状关键中间体13,不经纯化直接进行下一步反应。ESI-HRMS(m/z): 395.971 6 [M+H]+。1H NMR (400 MHz, DMSO-d6)δ: 11.51(s, 1H), 8.87(s, 1H), 8.60(s, 1H), 7.67(dd,J=8.8, 1.7 Hz, 1H), 7.62~7.58(m, 1H), 7.49(s, 1H), 4.02(s, 3H), 2.39(s, 3H)。
2.2.3 4-((3,4-二氯-2-氟苯基)氨基)-7-甲氧基喹唑啉-6-醇(14)
将中间体13(25.0 g,63.3 mmol)和甲醇(250 mL)置于1 000 mL反应瓶中,加入无水碳酸钾(26.2 g,189.9 mmol),室温下搅拌2 h,TLC检测反应完全(展开剂:二氯甲烷-甲醇,体积比10∶1),减压蒸除溶剂,将残留物倒入水(300 mL)中,抽滤,用水洗涤滤饼,滤饼经干燥后得到黄色固体(14)(21.5 g,两步收率96%)。ESI-HRMS(m/z): 354.053 2[M+H]+。1H NMR(400 MHz, DMSO-d6)δ:8.28(s,1H),7.61~7.54(m,2H),7.50(d,J=9.0 Hz,1H),7.14(s,1H),3.92(s,3H)。
2.2.4 4-((4-((3,4-二氯-2-氟苯基)氨基)-7-甲氧基喹唑啉-6-基)氧基)哌啶-1-羧酸叔丁酯(9)
将中间体14(20.0 g,56.7 mmol)和DMF(200 mL)置于1 000 mL反应瓶中,加入4-(甲苯-4-磺酰氧)哌啶-1-羧酸叔丁酯(化合物11,24.1 g,68.0 mmol)、无水碳酸钾(23.5 g,170.0 mmol),室温反应过夜,TLC检测反应完全(展开剂:二氯甲烷-甲醇,体积比10∶1),将混合物减压蒸除溶剂,得到黄色固体(9),加入二氯甲烷(30 mL)和甲醇(30 mL)的混合溶剂重结晶,得到化合物(9)纯品(20.1 g,收率66%)。ESI-HRMS(m/z): 537.146 8[M+H]+。1H NMR(400 MHz, DMSO-d6)δ:9.63(s,1H),8.39(s,1H),7.87(s,1H),7.60(d,J=5.9 Hz,2H),7.24(s,1H),3.94(s,3H),3.69(d,J=13.3 Hz,2H),1.64(d,J=11.9 Hz,2H),1.42(s,9H),1.26~1.22(m,3H),0.84(d,J=7.3 Hz,1H)。
2.2.5 N-(3,4-二氯-2-氟苯基)-7-甲氧基-6-(哌啶-4-氧基)喹唑啉-4-胺(10)
将中间体9 (11.0 g,20.5 mmol)置于500 mL反应瓶中,滴加二氯甲烷(DCM)和三氟乙酸(体积比4∶1)的混合溶液(110 mL),室温反应4 h,TLC检测反应完全(展开剂:二氯甲烷-甲醇,体积比10∶1)。减压蒸除溶剂,所得残留物残渣在冰浴下滴加饱和碳酸氢钠水溶液(100 mL)调pH值至8.0~9.0,随后用二氯甲烷进行萃取(3×150 mL),合并有机层,用无水硫酸钠干燥,减压蒸除溶剂得到黄色固体(10)(7.7 g,收率86%)。ESI-HRMS(m/z):437.094 6[M+H]+。1H NMR (400 MHz, DMSO-d6)δ:9.73(s,1H),8.38(s,1H),7.86(s,1H),7.59(d,J=5.2 Hz,2H),7.23(s,1H),4.64(tt,J=8.5,3.8 Hz,1H),3.95(s,2H),3.18(s,1H),3.06(dt,J=12.5,4.6 Hz,2H),2.75~2.68(m,2H),2.05(td,J=8.0,3.8 Hz,2H),1.66~1.56(m,2H) 。
2.2.6 Poziotinib(1)
在250 mL三颈瓶中将中间体10(4.7 g,10.8 mmol)溶于N,N-二甲基甲酰胺(50 mL),在0 ℃和氮气保护下,并依次缓慢滴加三乙胺(3.5 g,21.6 mmol)、丙烯酰氯(1.9 g,21.3 mmol)的二氯甲烷(20 mL)溶液,室温下搅拌进行反应12 h,TLC检测反应完全(展开剂:二氯甲烷-甲醇,体积比10∶1)。将反应液滴进水(200 mL)中,固体析出,抽滤,滤饼用水(100 mL)洗涤,真空干燥(45 ℃,12 h)得到灰白色固体(4.9 g)。常温下用二氯甲烷(6 mL)和甲醇(4 mL)溶解粗品,再滴加乙酸乙酯(12 mL)搅拌8 h,析出固体。抽滤,用乙酸乙酯(10 mL)洗涤,真空干燥(45 ℃,24 h)得到黄色固体(1)(4.4 g,收率84%),HPLC纯度为99.46%(HPLC面积归一化法),色谱柱:Ultimate C18柱(4.6 mm×250 mm,5 μm);流动相A:0.1%(体积比)的三氟乙酸水溶液;流动相B:乙腈;等度洗脱(B 70%)15 min;检测波长:254 nm;流速:0.8 mL·min-1;柱温:40 ℃。样品用乙腈溶解配置成浓度为100 μg·mL-1的溶液1 mL,进样量为10 μL。ESI-HRMS(m/z): 490.097 5[M+H]+。1H NMR (400 MHz, DMSO-d6)δ: 9.71(s,1H),8.40(s,1H),7.90(s,1H),7.60(d,J=5.5 Hz,2H),7.25(s,1H),6.85(d,J=6.1 Hz,1H),6.14~6.10(m,1H),5.71~5.67(m,1H),4.80~4.76(m,1H),3.95(s,3H),3.89(d,J=3.7 Hz,2H),3.54~3.46(m,2H),2.02(d,J=11.7 Hz,2H),1.73~1.66(m,2H)。
3 结果与讨论
3.1 起始原料的替换
本研究对工艺路线的起始物料进行重新核算,发现以5为起始原料更合适,使得反应路线由原文献的9步缩短为6步,减少了制备步骤,节约了制备成本,也大大提高了收率(表1)。

表1 路线收率Tab. 1 The yield of each routes
3.2 合成方法优化
3.2.1 化合物6的合成方法优化
合成化合物6时,原研路线选择三氯氧磷为氧化剂,甲苯作为溶剂,甲苯作为易制毒、易制爆试剂,在工业生成中可能无法大量使用,故优化路线后选择中等毒性的氯化亚砜作为氯化试剂和反应的溶剂。
3.2.2 脱乙酰基方法优化
合成中间体7时,原研路线选择以甲醇作为溶剂,氨水作为碱解试剂,但氨水在使用过程中,易挥发且有强烈的刺鼻气味,不适合工业大规模使用。本研究采用无水碳酸钾进行碱解,室温反应2 h后直接抽滤并用甲醇洗涤滤饼,滤液经浓缩、干燥后得到化合物14,收率可达95.7%。采用碳酸钾对乙酰基进行碱解,操作更为简单,更为安全,符合工业生产的要求。两种方法对比如表2所示。

表2 不同试剂的脱乙酰条件和产率Tab. 2 Deacetylation conditions and yields of different reagents
3.2.3 中间体10的方法优化
合成中间体10时,原研路线脱除Boc保护基时,以丙酮作为溶剂。本研究采用三氟乙酸和二氯甲烷体系进行Boc脱除,在不降低收率的同时,提高了反应的安全性。丙酮作为易制毒试剂,成本较高,据核算,吨位级丙酮价格约为二氯甲烷的1.5倍,且在研发和生产中使用颇为不便。本研究的方法可为相关化合物的脱保护提供一种参考方案。
3.2.4 中间体9和终产物纯化方法优化
在纯化中间体9和终产物时,本工艺路线经实验摸索出合适的重结晶纯化方法,对得到的化合物进行重结晶纯化,避免了使用柱层析等纯化方法,且所选择的重结晶溶剂为工业常用品且价格低廉,更适用于工业放大生产。
4 结论
作者改进了Poziotinib的合成路线,以价格便宜的化合物5为起始原料,用氯化亚砜作为氯化原料代替三氯氧磷和甲苯,更符合工业生产要求。中间体13经过取代后以无水碳酸钾代替氨水作为酯水解反应试剂,得到中间体14,收率得到了提高。中间体14与4-(甲苯-4-磺酰氧)哌啶-1-羧酸叔丁酯(11)酯化、脱保护基得中间体9,再与丙烯酰氯经酰化反应得到目标产物Poziotinib(1),改进后路线总收率为43.2%,该方法合成步骤短、后处理简单,无需复杂的纯化即可进行下一步反应,且使用的试剂更适合工业放大生产,可为Pozionitib及其衍生物的合成提供参考。
