泥鳅黏液多糖的化学组成、理化性质及体外降糖活性
2023-12-05李亚楠郭明珠邵娟娟桑亚新孙纪录
李亚楠,郭明珠,邵娟娟,桑亚新,孙纪录*
(1 河北农业大学理工学院 河北沧州061100 2 河北农业大学食品科技学院 河北保定 071000)
大鳞副泥鳅(Paramisgurnus dabryanus)属鲤形目、鳅科、花鳅亚科、副泥鳅属,主要分布于长江、嘉陵江和岷江水系、辽河中下游、黄河及黑龙江等地区,是一种营底栖生活的小型鱼类[1]。其营养价值高,具有极快的生长速度和较高的经济价值,近年来,逐渐成为广大养殖户选择的品种之一[2]。
泥鳅体表能分泌出大量黏液,尤其是在冷链运输和加工预处理过程中。泥鳅黏液入药味甘、性平,具有补中益气、利尿除湿等功效[3]。鱼皮的黏液层含有各种来自表皮杯状细胞和上皮细胞的分泌物,这些分泌物与许多重要的生物学功能有关[4]。泥鳅黏液中含有超氧化物歧化酶、抗菌肽、多糖等多种生物活性物质[5]。目前,在泥鳅运输和加工过程中产生的大量黏液被废弃,不仅造成极大的资源浪费,还带来环境污染问题。
泥鳅黏液多糖作为一类生物活性物质,已有少数研究报道。Qin 等[6]从普通泥鳅(Misgurnus anguillicaudatus)黏液中分离纯化出一种分子质量为1.30×105u 的泥鳅多糖(Misgurnus anguillicaudatus polysaccharide,MAP),该多糖具有很强的抗氧化能力、免疫活性和恢复肝脏损伤等功能。孙智华等[7]利用泥鳅黏液制备出一种分子质量为1.57×104u 的具有保湿性的透明质酸,在化妆品和医药行业具有应用价值。Kimura 等[8]从泥鳅黏液中分离并鉴定出一种含神经氨酸的脱胺糖蛋白。孙朋朋[9]从泥鳅黏液中分离出含有O-型糖基化结构的凝集素。目前,关于泥鳅的研究报道主要集中于养殖和遗传方面,对泥鳅黏液中的多糖研究较少,而且主要是针对普通泥鳅的黏液。对大鳞副泥鳅的黏液多糖及其抗糖活性鲜有报道。
本文从泥鳅副产物资源的综合利用角度出发,拟从大鳞副泥鳅黏液中分离多糖,对其化学组成、理化性质和结构进行研究,并探讨其体外抗糖活性,以期为大鳞副泥鳅黏液资源应用到食品和医药行业提供理论依据。
1 材料与方法
1.1 材料与仪器
1.1.1 材料与试剂 大鳞副泥鳅:购于河北农业大学科技市场。
葡萄糖、苯酚、三氯乙酸、酒石酸钾钠、3,5-二硝基水杨酸,福晨化学试剂有限公司;无水乙醇,天津市富宇精细化工有限公司;溴化钾,天津市科密欧化学试剂有限公司;浓硫酸、氢氧化钠、氯化钠为分析纯级,天津市津东天正精细化学试剂厂;牛血清白蛋白(BSA)、考马斯亮蓝G-250,北京索莱宝科技有限公司;甲醇、正丁醇,国药集团化学试剂有限公司;三氟乙酸,上海安普实验科技股份有限公司。
1.1.2 设备与仪器 TGL21M 台式高速冷冻离心机,长沙易达仪器有限公司;YTLG-12A 台式真空冷冻干燥机,上海叶拓科技有限公司;721G 可见分光光度计,上海仪电分析仪器有限公司;Multiskan Spectrum 酶标仪,Thermo scientific 公司;高效液相(HPLC)色谱仪(LC-20A),日本岛津公司;核磁共振波谱仪(Bruker 400M),德国Bruker 公司;Nicolet iS50 型傅里叶变换红外光谱仪(FTIR),美国Thermo Fisher Scientific 公司;X 射线衍射仪(日本理学Ultima IV),热重分析仪(NETZSCH STA 409 PC/PG),耐驰科学仪器商贸有限公司。
1.2 试验方法
1.2.1 PDMP 的化学组成分析
1.2.1.1 PDMP 的制备 鲜活泥鳅→蒸馏水浸没暂养12 h→收集黏液→浓缩→超声→醇沉→去蛋白→透析→冷冻干燥→泥鳅黏液多糖。
1.2.1.2 PDMP 的成分测定 采用苯酚硫酸法测定总糖含量[10],3,5-二硝基水杨酸法测定还原糖含量[11],考马斯亮蓝法测定蛋白质含量[12],采用BaCl2-明胶法测定硫酸基含量[12],采用咔唑硫酸法测定糖醛酸含量[12]。
1.2.1.3 紫外光谱分析 使用紫外(UV)分光光度计扫描,将PDMP 样品制备1 mg/mL 的去离子水溶液[13],并在200~400 nm 波长范围内记录光谱。
1.2.1.4 分子质量测定 根据文献采用HPGPC法[14]测定PDMP 的平均分子质量,以葡聚糖T-10,T-40,T-70,T-500 和T-2000 葡聚糖为分子质量标准品,测定获得 “分子质量-保留时间标准曲线”。
1.2.1.5 单糖组成
1)样品提取
a)固体样本提取 取干净的色谱瓶,精确称量多糖样品5 mg,加入1 mL 2 mol/L TFA 酸溶液,105 ℃加热6 h。通氮气,吹干。加入甲醇清洗,再吹干,重复甲醇清洗2~3 次。加入无菌水溶解,转入色谱瓶中待测。
b)液体样本提取 取适量的上清液,旋转浓缩或氮气吹干。加入1 mL 2 mol/L TFA 酸溶液,105 ℃加热6 h。通氮气,吹干。加入甲醇清洗,再吹干,重复甲醇清洗2~3 次。加入无菌水溶解,转入色谱瓶中待测。色谱系统采用的是Thermo ICS5000 离子色谱系统(ICS5000,Thermo Fisher Scientific,USA),利用电化学检测器对单糖组分进行分析检测。
2)仪器参数 采用DionexTMCarboPacTMPA20(150 mm×3.0 mm,10 μm)液相色谱柱;进样量为5 μL。流动相A(0.1 mol/L NaOH),流动相B(0.1 mol/L NaOH,0.2 mol/L NaAc),流速0.5 mL/min;柱温为30 ℃;洗脱梯度:0 min A 相/B 相(95∶5,V/V),30 min A 相/B 相(80∶20,V/V),30.1 min A 相/B 相(60∶40,V/V),45 min A 相/B 相(60∶40,V/V),45.1 min A 相/B 相(95 ∶5,V/V),60 min A相/B 相(95∶5,V/V)。
1.2.1.6 傅里叶变换红外光谱 红外光谱(IR)测定:将PDMP 和KBr 按质量比1∶100 混合研磨均匀(提前于105 ℃的烘箱中烘干),并压成1 mm 薄片在4 000~500 cm-1范围内扫描,测定多糖的有机官能团。
1.2.1.7 核磁共振波谱(NMR)测定 PDMP 的1H NMR 和13C NMR 的测定:取20 mg PDMP 溶解在1 mL 氧化二氘(D2O)中,使用400 MHz 傅立叶变换核磁共振波谱仪。在25 ℃下记录1H 和13C的NMR 光谱[10]。
1.2.2 理化性质分析
1.2.2.1 热稳定性测定 采用STA449C 型同步热分析仪,测定PDMP 的热稳定性能[15]。取10 mg 样品,测试条件:温度范围30~700 ℃,升温速率(β)5℃/min,氮气气氛,气体流量20 mL/min。
1.2.2.2 结晶度测定 采用X-射线衍射仪(XRD)测定PDMP 的结晶度[16]。XRD 在散射角范围(2θ,10°~80°)下进行,步长和曝光时间分别为0.05 s 和1 s。
1.2.2.3 微观结构观察 参照何坤明等[17]的方法,将样品干燥至恒重,取适量进行黏台、镀金后,使用HitachiS-4800 扫描电子显微镜在放大500 倍和2 000 倍观察PDMP 表面的微观形态结构。
1.2.3 体外降血糖活性
1.2.3.1 α-葡萄糖甘酶抑制活性测定 α-葡萄糖甘酶的活性测定根据Getachew 等[16]的方法并略作改动,将PDMP 分别配制成不同浓度梯度的多糖溶液(1.0,3.0,5.0,7.0,9.0 mg/mL),分别取400 μL的样品与50 μL α-葡萄糖甘酶(0.2 U/mL)混合,在37 ℃下反应10 min,反应所用缓冲溶液为0.1 mol/L,pH 5.5 的磷酸盐缓冲溶液,然后加入1 mg/mL 还原型谷胱甘肽和PNPG,分别为50 μL 和100 μL,在37 ℃下反应30 min 后,通过加入1 mL NaCO3(0.1 mol/L)终止催化反应。以阿卡波糖为阳性对照。α-葡萄糖甘酶抑制率计算方法:
式中,As——样品组的吸光值;Ab——阴性空白对照组(不含样品和酶)的吸光值;Ac——样品对照组(不含酶)的吸光值。
1.2.3.2 α-淀粉酶抑制活性的测定 将100 μL α-淀粉酶溶液和不同浓度的多糖溶液混合并在25 ℃下反应10 min,然后加入可溶性淀粉溶液100 μL,继续反应10 min,随后加入200 μL 3,5-二硝基水杨酸并在沸水中加热5 min,然后将混合物冷却至室温并用3 mL 去离子水稀释。在540 nm 波长处测定吸光度,以阿卡波糖为阳性对照。α-淀粉酶抑制率计算方法:
式中,As——样品组的吸光值;Asc——样品对照组(不含酶)的吸光值;Ac——阴性对照组(不含样品)的吸光值;Abc——阴性空白对照组(不含样品和酶)的吸光值。
1.3 数据处理
3 次重复试验的结果以 “平均值±标准差”表示,使用Microsoft Office ExceL 2010 处理数据,数据运用SPSS 19.0 进行统计分析,Origin 2018软件绘图。
2 结果与分析
2.1 PDMP 的化学组成分析
2.1.1 PDMP 中的主要化学成分 表1 显示了PDMP 的化学成分。

表1 PDMP 的主要化学成分Table 1 Main chemical constituents of PDMP

表2 PDMP 的凝胶过滤色谱分析Table 2 Analytical results of PDMP by GPC
如表1 所示,PDMP 含有(2.87±0.09)%的蛋白质和(95.51±2.27)%的总糖,其还原糖含量为(13.28±0.10)%,硫酸基含量为(23.41±0.87)%,不含糖醛酸。以上研究结果表明制备的PDMP 纯度较高,可以用于后续的测定。此外,多糖中含有大量的硫酸基,表明可能与多种功能活性相关,例如抗肿瘤、抗氧化、抗凝血等活性,有研究表明硫酸基与降血糖功能存在一定相关性,可进一步研究其降血糖活性。
2.1.2 PDMP 的紫外光谱 PDMP 通过紫外-可见光谱进行检测,吸收光谱如图1 所示。

图1 PDMP 的UV 光谱Fig.1 UV spectra of PDMP
由图1 可以看出,在200 nm 波长处存在多糖的特征吸收峰,PDMP 在260~280 nm 处无明显吸收峰,表明不存在核酸类和蛋白类杂质,说明PDMP 的纯度较高,而在280 nm 波长处,只出现微弱吸收峰,表明PDMP 经脱蛋白处理后,有极少量蛋白与多糖紧密结合[18],这与Bradford 法测定蛋白质含量的结果一致,说明去除蛋白的效果明显。
2.1.3 PDMP 的分子质量 多糖的分子质量被证实是影响多糖的理化和生物活性差异的重要指标[14]。
PDMP 的高效凝胶渗透色谱(HPGPC)的测定结果显示,PDMP 的主要组分的重均分子质量为362 928 u,数均分子质量为237 749 u。多分散性指数(Mw/Mn)为1.527,接近1,说明PDMP 的平均分子质量分布较集中。PDMP 的Mw 为362 928 u。分子质量的表征有助于进一步研究其与活性的关系,并有助于了解其潜在的应用价值。
2.1.4 PDMP 的单糖组成 如图2a 所示混合标准品中各单糖的出峰时间依次为3.692,7.584,7.892,9.95,11.392,13.392,14.125,16.509,18.067,34.192,34.867,36.725,38.992 min。

图2 单糖标准品(a)和PDMP(b)的离子色谱图Fig.2 Ion chromatograms of standard substance(a)and PDMP(b)
根据单糖标准品衍生化的保留时间鉴定PDMP 的单糖组成,如图2b 所示,PDMP 的单糖组成主要由岩藻糖、半乳糖、甘露糖、葡萄糖和木糖组成,其物质的量比为13.27 ∶5.68 ∶2.31 ∶2.23 ∶0.45。根据已发表的泥鳅黏液多糖的文献表明,单糖组成有一定差异,已经报道的泥鳅黏液多糖的单糖组成的主要结构单体由半乳糖、岩藻糖和甘露糖(5∶4∶1)组成[19];本文中PDMP 主要组分岩藻糖和半乳糖,所占百分比分别为55.42%和23.74%,甘露糖、葡萄糖和木糖所占百分比分别为9.66%,9.33%,1.86%。这可能是由于原料的种类、产地及生活习性不同导致。以上结果表明,PDMP 主要为岩藻半乳糖,此外FT-IR 光谱分析中有α-型糖苷键的信号,因此单糖组分中的糖有可能存在α 异构型。
2.1.5 PDMP 的FT-IR 光谱分析 FT-IR 分析是确定多糖基本结构特征的经典方法。FT-IR 光谱的指纹区域是基于对特定波数吸收峰的分析[20],官能团分析基于范围从4 000~1 333 cm-1的特征吸收峰面积,1 000 cm-1以下的指纹区域提供了更多关于复杂结构的信息[21]。
如图3 所示,在3 423 cm-1附近的特征性强宽带表明氢键中存在O-H 拉伸[22],表明多糖链的分子间和分子内相互作用很强。2930 cm-1和1400~1 200 cm-1处分别对应C-H 伸缩振动和C-H 变角振动[23];1 642 cm-1和1 554 cm-1处的双峰归因于-CH2-CO-中的C=O 的伸缩振动及PDMP 中的结晶水[24-25];在1 188,1 118 cm-1和1 060 cm-1处的吸收峰是吡喃环骨架的C-O 变角振动吸收峰[26],表明分子中存在C-O-H 和C-O-C 结构[27];在1 060 cm-1和615 cm-1处为硫酸根的伸缩振动峰[26];出现在884 cm-1附近的吸收峰,表明吡喃糖的存在[28],具体的连接键型(α-,β-)需要后续的核磁共振验证。从上述结果可以推断PDMP 有可能为含有硫酸根的硫酸化多糖。由于多糖的天然特性,不同糖残基产生的一些信号可能在指纹区域重叠,然而,大多数典型信号可以根据准确的单糖组成分析和NMR 谱图进行分析比对。

图3 PDMP 的FT-IR 光谱Fig.3 FT-IR spectrum of PDMP
2.1.6 PDMP 的NMR 光谱分析 通过1H 和13C NMR 光谱以进一步研究糖基残基的连接,PDMP的1H NMR 和13C NMR 图,如图4 和5 所示。
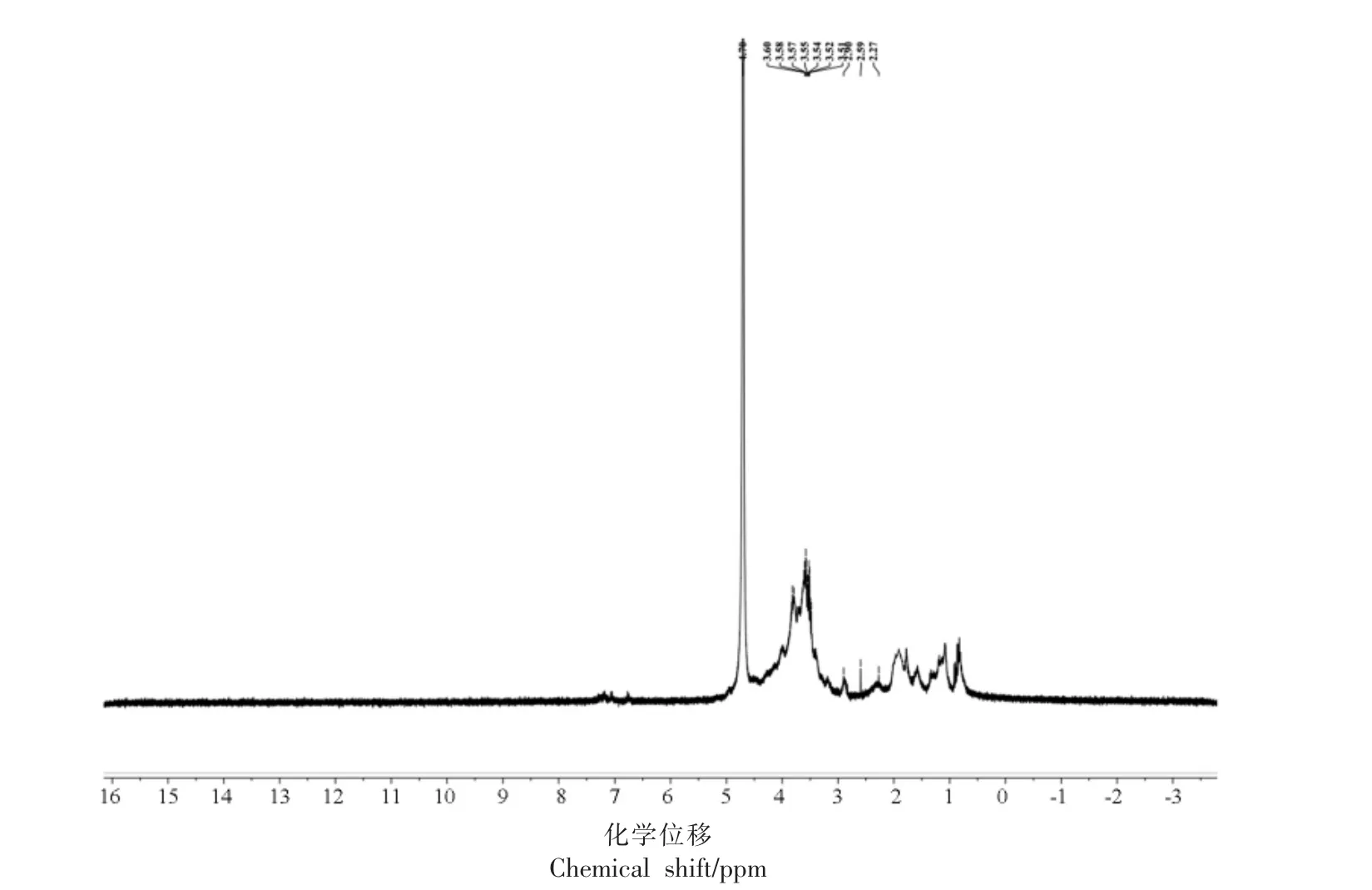
图4 PDMP 的1H NMR 光谱图Fig.4 1H NMR spectra of PDMP
1)PDMP 的1H NMR 光谱
在PDMP 的1H NMR 谱图中,多糖中糖残基的异头质子的构型可以根据它们的化学位移(δ)从它们的1H-NMR 光谱中识别出来。一般来说,4.9~6.0 ppm 范围内的异头质子是α-构型,而4.0~4.9 ppm 范围内的异头质子是β-构型[29]。在1H NMR 谱图中,PDMP 在δ 4.0~4.9 ppm 处的吸收峰,表明了异头质子的存在,δ H 大约在3.4~4.0 ppm 的值代表了多糖中的非异头原子H2-H6。在δ 5.0~5.3 ppm 并未出现异头质子信号,说明PDMP 中不存在α-Galp A 残基的异头氢质子信号[30]。在δ 1.8~2.3 ppm 的共振信号归属于乙酰基(CH3CO-)的甲基质子信号,为GalNAc 中甲基的(CH3-)[31],进一步印证了FT-IR 和单糖组成的分析结果。
2)PDMP 的13C NMR 光谱 与1H-NMR 相比,13C-NMR 化学位移范围宽广,分辨率高,可以解析异头碳的构型,多糖残基中取代位点和分支点[32]。13C NMR 谱中异头碳的共振峰位于δ 95.0~110.0 ppm 范围内。在PDMP 的13C NMR 谱图中,异头碳 的化学 位移有100.35,102.10,103.19 ppm,表明存在α-糖苷键和β-糖苷键构型[33],另一方面,吡喃糖的C-3 和C-5 的化学位移一般小于80 ppm,而呋喃糖一般介于82~84 ppm 之间[34]。图中可以看出,多个碳的信号峰,除了异头碳的信号外,其它均小于80 ppm,因此判断PDMP 为吡喃糖,这也与上文红外光谱测定的结果相一致。δ C大约在60~90 ppm 的信号峰,代表了多糖中的非异头原子C2-C6,13C-NMR 图中δ 71.34 ppm 和δ 63.28 ppm 区域的峰分别对应于C2-C5 和C-6 的峰信号[35],60~70 ppm 之间有化学位移,表明C6 位置羟基一部分发生取代,C2-C5 信号的δ 值基本全部集中在70~77 ppm 之间,表明其绝大部分未被取代,还有一小部分被取代[36],而图5 中δ 75~85 ppm 没有峰信号,因此,C2-C5 位置上的羟基没有被取代。在δ 27.01 ppm,29.97 ppm 和δ 39.74 ppm 处的碳信号均在小于78 ppm,进一步证实PDMP 的糖环为吡喃糖环[37]。δ 174.56 ppm的峰信号乙酰基的存在[38],证实了FT-IR 的分析结果,表明PDMP 可能为一种硫酸化多糖。由1H NMR 和13C NMR 的核磁信息,PDMP 含有(1→)-和(1→6)-糖苷键的α-糖基和β-糖基。党超[11]从泥鳅鱼头中提取的泥鳅多糖中同时存在α 型吡喃糖和β 型吡喃糖,这与本研究的PDMP 相似。

图5 PDMP 的13C NMR 光谱图Fig.5 13C NMR spectra of PDMP
2.2 PDMP 的理化性质分析
2.2.1 PDMP 的热稳定性 图6 为PDMP 热降解质量变化与变化速率曲线。TG 是样品质量随温度变化曲线,DTG 是TG 的一阶微分曲线,表示样品失重速率与温度的关系,通过热重分析可以了解样品的热稳定性。
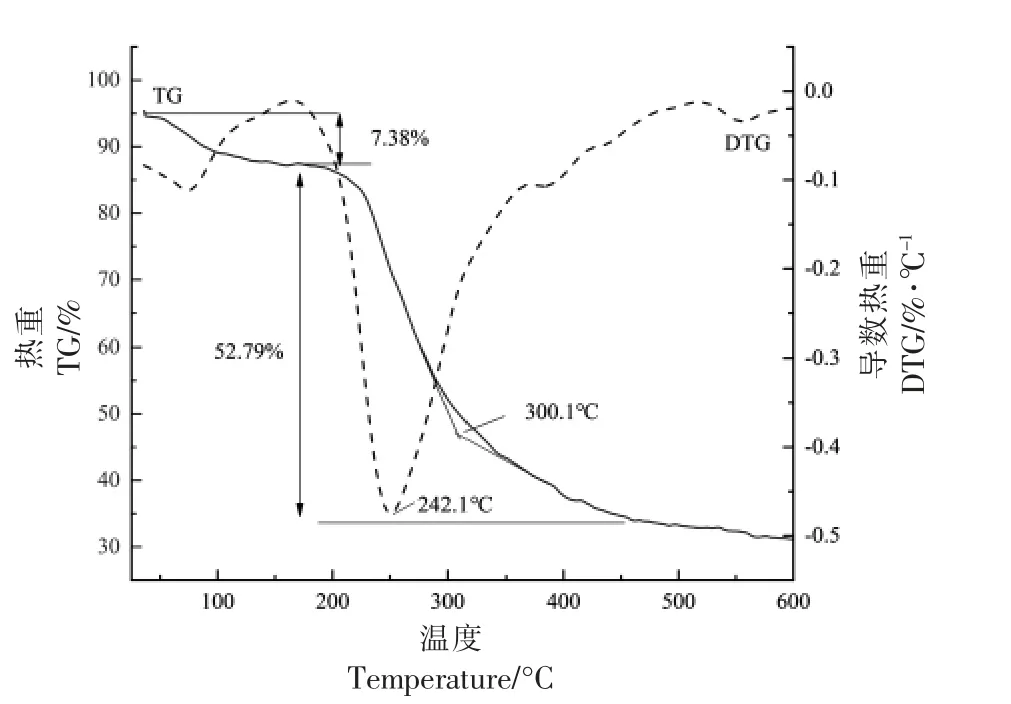
图6 PDMP 的TG 曲线(a)和DTG 曲线(b)Fig.6 TG curve(a)and DTG curve(b)of PDMP
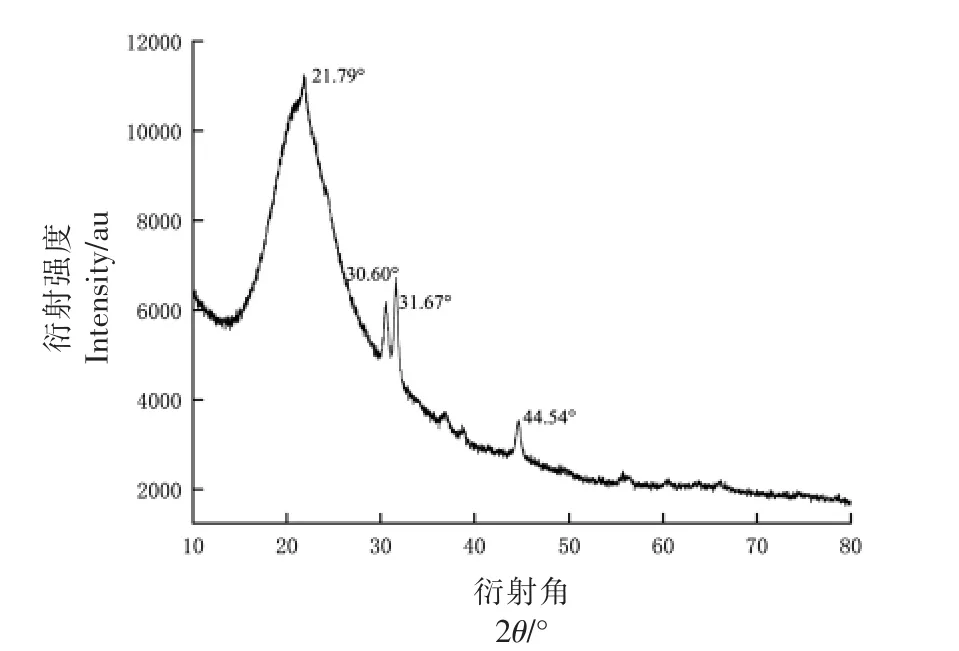
图7 PDMP 的X-射线衍射图Fig.7 X-ray diffraction pattern of PDMP
TG 结果显示随着温度的变化PDMP 的质量损失率,在200 ℃之前PDMP 并没有因为升温而出现明显的失重,质量损失仅为7.38%,这部分的质量损失主要是由于多糖物理吸附水的挥发[37]。当温度超过200 ℃时,PDMP 出现明显的质量损失,损失率达52.79%,由DTG 曲线可以看出,多糖在300 ℃之前降解速率很快,242.10 ℃时降解速率达到最快,300 ℃之后降解开始逐渐降低,在200 ℃到400 ℃范围内有较大的质量损失,可能是分子质量相对较大或稳定性高的成分在这个阶段温度内被降解[38]。400 ℃时开始趋于平缓,到达终末温度600 ℃时,物质并没有完全分解,PDMP 质量残留率为31.25%,主要是残余物成分慢慢碳化阶段,还与质量损失中的一些未分解物质有关[39]。该结果表明,PDMP 的热稳定性较高,图8 PDMP的扫描电镜图显示比较规则的排列,分子间相互作用力较强,可能这种物理结构使其更不易热分解。

图8 PDMP 的扫描电镜图Fig.8 SEM images of PDMP
2.2.2 PDMP 的结晶性 X 射线衍射是确定物相、晶体结构和其它结构参数的有用工具[40]。通过X 射线衍射分析PDMP 的结晶性能。
根据PDMP 的XRD 图,可以看出,PDMP 的衍射角在30.60°,31.67°和44.54°有明显尖峰,说明其含有粒度较大的微晶[41];在21.79°时有弥散宽尖峰,说明其不仅存在微晶体系,还存在微晶与非晶共存、亚微晶与非晶共存的多晶体系[43]。这与蒋茂婷等[42]提取的蒜皮水溶性多糖的结晶性有相似之处。以上结果表明PDMP 的可塑性高,PDMP 是一种很有前途的功能性高分子碳水化合物。
2.2.3 PDMP 的表面形貌观察 经脱蛋白处理后的PDMP 的表面形貌通过SEM 在500 倍和2 000倍放大倍数下观察。
如图8 所示,样品分别以100 μm 和20 μm标尺拍摄图片,PDMP 的表面形成大小均一密集的蜂窝状孔洞,内部有大小不一的孔洞结构,说明PDMP 的成分相对均一,分子间相互作用力较强。
2.3 PDMP 的体外降糖活性评价
2.3.1 PDMP 的α-淀粉酶抑制活性 α-淀粉酶是一种碳水化合物降解酶,它最初作用于高分子质量的糖类,并将它们分解成较小分子质量的低聚糖,然后通过α-葡萄糖苷酶进一步将低聚糖降解为单糖,随后导致血液中的糖水平升高[17]。
如图9 所示,在不同浓度下,PDMP 对α-淀粉酶都有抑制活性,在1~9 mg/mL 范围内,PDMP 对α-淀粉酶的抑制活性呈一定的剂量依赖关系,相同浓度下PDMP 的抑制效果要低于阳性对照组阿卡波糖。PDMP 和阿卡波糖的50%抑制浓度(IC50)分别为152 μg/mL 和476 μg/mL。当质量浓度为9 mg/mL 时,PDMP 和阿卡波糖的抑制率分别达到65.02%和88.70%。Lakshmanasenthil 等[43]发现喇叭藻(Turbinaria ornate)中褐藻糖胶硫酸基含量为(33.00±0.42)%,具有较强的α-淀粉酶抑制作用,IC50值为33.60 μg,低于阿卡波糖125 μg。本研究推测PDMP 中硫酸基的存在是其抑制其活性的原因之一,可进一步深入研究其构效关系。

图9 PDMP 和阿卡波糖的α-淀粉酶抑制活性Fig.9 α-Amylase inhibitory activity of PDMP and acarbose
2.3.2 PDMP 的α-葡萄糖苷酶抑制活性 降低餐后高血糖的α-葡萄糖苷酶抑制剂可以调节血糖水平减缓糖尿病的进展,是治疗Ⅱ型糖尿病前期状态的关键因素[18]。PDMP 体外对α-葡萄糖苷酶的抑制活性如图10 所示。
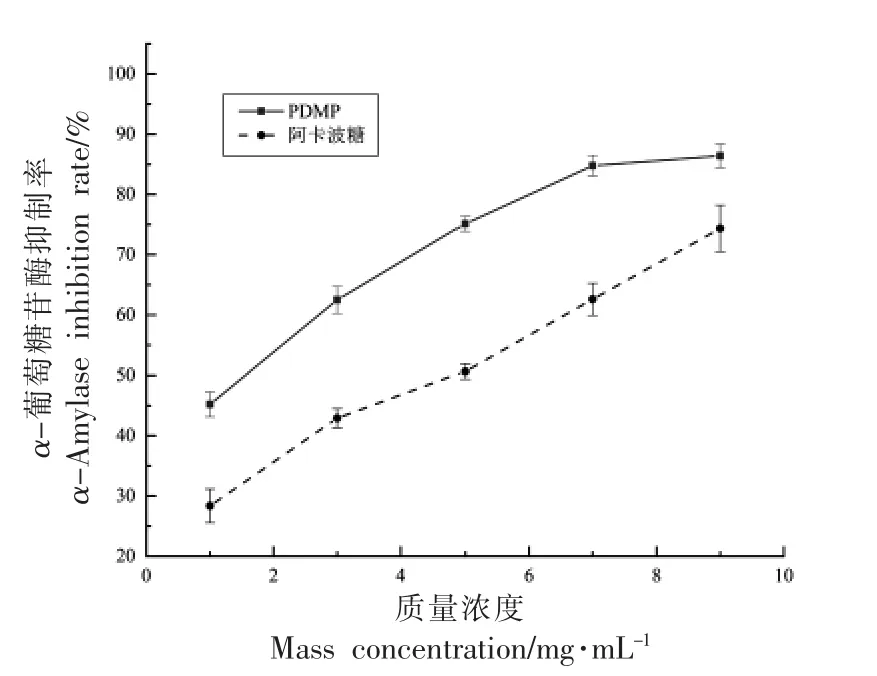
图10 PDMP 和阿卡波糖的α-葡萄糖苷酶抑制活性Fig.10 α-Glucosidase inhibitory activity of PDMP and acarbose
如图所示,在不同浓度下PDMP 对α-葡萄糖苷酶有抑制活性,在1~9 mg/mL 范围内,PDMP 对α-葡萄糖苷酶的抑制活性呈一定的剂量依赖关系,相同浓度下PDMP 的抑制效果要高于阳性对照组阿卡波糖。PDMP 和阿卡波糖的50%抑制浓度(IC50)分别为550,136 μg/mL。当质量浓度为9 mg/mL 时PDMP 和阿卡波糖的抑制率分别达到86.40%,74.35%。PDMP 表现出显著的对α-葡萄糖苷酶的抑制活性。Xiao 等[44]从马尾藻制备的硫酸化马尾藻多糖硫酸根含量为35.80%,对α-葡萄糖苷酶的抑制效果显著,在质量浓度为1 mg/mL时,抑制率达到98.40%,高于阿卡波糖。Zhong等[45]制备的硫酸化裙带菜多糖硫酸根含量为38.79%,IC50值为50.50 μg/mL,比阿卡波糖更有效(IC50值为337.30 μg/mL)。以上结果说明含有硫酸基的天然PDMP 对α-葡萄糖苷酶抑制活性较高,猜测可能与其本身含有大量硫酸根有一定关系,此外,含有较多的岩藻糖和半乳糖以及阿拉伯糖和木糖都具有显著的α-葡萄糖苷酶活性,因此,PDMP 可能是缓解Ⅱ型糖尿病的一种潜在物质,为后续深入研究其机制奠定理论基础。
3 结论
从大鳞副泥鳅黏液中分离出一种热稳定较强,有一定结晶性的多糖-PDMP,其总糖含量为(95.51±2.27)%,富含硫酸基,不含糖醛酸,Mw 为362 928 u。PDMP 是由岩藻糖、半乳糖、甘露糖、葡萄糖、木糖5 种单糖组成的岩藻半乳糖,其摩尔比为13.27∶5.68∶2.31∶2.23∶0.45。PDMP 含有(1→)-和(1→6)-糖苷键的α-糖基和β-糖基的吡喃糖。此外,PDMP 在体外对α-淀粉酶和α-葡萄糖苷酶活性的抑制效果良好,展现出一定的降糖潜力。这些研究结果为PDMP 在医药领域和现代食品工业的应用奠定了一定的理论基础。
