基于GC-MS/MS 法测定鱼肉中二噁英和二噁英类多氯联苯
2023-11-26陆静申甜甜焦艳娜崔凤云朱绍华成婧易锡付善良
陆静,申甜甜,焦艳娜,崔凤云,朱绍华,成婧,易锡,付善良*
(1 长沙海关技术中心 食品安全科学技术湖南省重点实验室 长沙410004 2 中国海关科学技术研究中心 北京 100026)
二噁英类(Dioxins,DXNs)污染物是一类具有相似化学结构和理化性质及生物效应的多氯代三环芳烃类化合物的统称,在结构上分别为多氯代二苯并对二噁英(PCDDs,75 种异构体),氯代二苯并呋喃(PCDFs,135 种异构体)和共平面多氯联苯(Co-PCB)[1-2]。美国在越战中使用2,4-滴和2,4,5-涕等落叶剂所造成的危害后果及日本米糠油事件引发的灾难之后,人们开始认识到作为人类社会最为有毒、有害的一类化学物质,DXNs 不仅具有难分解高生物蓄积性,还具有生殖毒性、神经毒性、内分泌毒性和免疫毒性效应等,被国际社会公认为环境内分泌干扰物[1,3-5]。其中,2,3,7,8-四氯代苯并二噁英(TCDD)是迄今为止发现毒性最强的化合物,比氰化钾要毒100 倍以上,被世界卫生组织判定为一级致癌物。二噁英类物质毒性的另一个特点是,二噁英为脂溶性化合物,易积累在生物体的脂肪组织中,不易被降解和排出,可在人和动物体内不断蓄积达到较高浓度,在生物体表现出明显的症状之前有一个漫长的潜伏过程[6-9]。目前,国内还没有二噁英类物质的限量标准,根据(EC)No 1881-2006 欧盟食品污染物限量标准[10],食品中二噁英总和的最高限量值为6 pg/g,多氯联苯的最高限值为12 pg/g。
DXNs 分析属于超痕量、多组分分析,其分析测定必须具备有效的采样技术,定量提取和净化技术,异构体的高效分离,准确的定性和定量,以及良好的质量管理等技术条件[11-12]。且同位素稀释内标法是国际上通用的测定食品、饲料及环境样品中二噁英质量浓度的方法[13-15]。在食品安全受到广泛关注下,我国修改并采用了美国EPA 1613和EPA 1668A,提出适合我国食品中二噁英以及类似物检测的标准[16]。在该项标准中,应用高分辨气相色谱-高分辨质谱联用(HRGC-HRMS)技术,在质谱分辨率大于10 000 的条件下,通过精确质量测量、监测目标化合物的两个离子,获得目标化合物的特异性响应。HRGC-HRMS 分析灵敏度高、抗基质干扰强,检出限可达到fg 级别。然而其运行和维护成本较高,对人员的操作水平也有较高要求。自2014 年6 月起,GC-MS/MS 成为欧盟委员会(EU)规程2017/644 中的一项确证方法[17]。目前,GC-MS/MS 法已广泛用于食品、饲料及环境样品中二噁英及二噁英类多氯联苯(PCDD/Fs 和DL-PCBs)的检测[18-21]。不少学者和专家对比研究了该两种仪器在检测PCDD/Fs 的区别。Palmiotto等[22]采集环境土样,研究了GC-MS/MS 和HRGCHRMS 测定同一份样品的处理后溶液中PCDD/Fs浓度的差异,结果发现在22 个样品中,PCDD/Fs的检出浓度为0.33~14.11 pg WHO-TEQ/g,当样品的PCDD/Fs 的最低浓度为1 pg WHO-TEQ/g时,两种仪器的测定偏差<20%;当样品的PCDD/Fs 的最低浓度为3 pg WHO-TEQ/g 时,两种仪器的测定偏差为<4%。Sun[23]对鱼、牛肉以及玉米饲料3 种基质中的PCDD/Fs 浓度进行GC-MS/MS 和HRGC-HRMS 仪器比对检测,检出浓度为0.9~9.0 pg/g,两种仪器的测定偏差为0.7%~14%。
综上所述,开发一种适合动物源性食品中PCDD/Fs 和DL-PCBs 的GC-MS/MS 方法是可行的[13,24]。目前发达国家均以此为技术堡垒,我国必须加快此类方法的标准化研究,建立国际认可的二噁英超痕量分析实验室,以应对分析技术的挑战[25-26]。该方法的建立和优化能够有力地保障关区动物源性食品的顺利出口。
1 材料与方法
1.1 试剂与仪器
二氯甲烷(农残级)、甲苯(农残级)、乙酸乙酯(农残级)、正己烷(农残级),iMagiLab 公司;硅胶(农残级),加拿大Silicycle 公司;浓硫酸(分析纯)、无水硫酸钠(分析纯),国药集团化学试剂有限公司;硅藻土,美国ThermoFisher 公司。多层硅胶柱、碱性氧化铝柱、活性炭柱,FMS 美国FMS 公司。
分离度检查标准溶液,同位素标记定量内标标准溶液、回收率内标标准溶液;含有天然和同位素标记的系列校正标准曲线标准溶液均从Wellington Laboratories(加拿大)公司购买,浓度同国家标准中规定[16],临用时取20 μL 于进样小瓶中,备用。
硅胶使用前先用甲醇和二氯甲烷活化,晾干后在600 ℃下至少烘烤12 h,再以浓硫酸酸化(44%)。
鱼肉样品为从当地市场购买的草鱼,去除内脏、鱼骨,肉样搅碎后以冷冻干燥机干燥,混匀备用。
气相色谱-三重四极杆串联质谱仪(Shimadzu GC-MSTQ8050 型),日本岛津公司;快速溶剂萃取仪(E-916)、旋转蒸发仪(R-300),瑞士步琦公司;全自动样品净化系统(JF-602),北京普立泰科公司;冷冻干燥机(Yamato DC801),日本雅马拓公司。
1.2 试验方法
1.2.1 样品前处理方法
1.2.1.1 提取 称取5.0 g 冷冻干燥后的样品于陶瓷研钵中,研磨后与适量硅藻土混合均匀,转移至40 mL 的萃取池中,加入PCDD/Fs 和DL-PCBs回收率和精密度检查标准溶液(B.4×10 和B.11×100)各50 μL,13C12-PCDD/Fs和13C12-DL-PCBs 标记的定量内标溶液(B.2 和B.8×10)各20 μL,密闭后置于加速溶剂萃取仪上提取,提取条件如下:提取溶剂正己烷:二氯甲烷(1∶1,体积比);压力10.3 MPa;温度150 ℃;第1 次、2 次、3 次循环静态提取时间分别为10,5,2 min;共计循环3 次。
1.2.1.2 除脂 将提取液转移至圆底烧瓶中,旋转蒸发浓缩至近干,加入100 mL 正己烷,50 g 酸化硅胶,用旋转蒸发仪在70 ℃条件下旋转加热20 min。静置8~10 min 后,将正己烷倒入圆底烧瓶中。残渣再用50 mL 正己烷洗涤,重复3 次,合并正己烷溶液,用旋转蒸发仪浓缩至2~5 mL。
1.2.1.3 净化 将多层硅胶柱、碱性氧化铝柱和活性炭柱按顺序接在全自动样品净化系统上,按程序配好各洗脱溶液并连接好管路,将上述试样转移到进样管中。按照洗脱程序顺序洗脱,对样品进行净化、分离,分别收集含PCDD/Fs 和DLPCBs 组分的洗脱液,用旋转蒸发仪浓缩至3~5 mL。
1.2.1.4 浓缩 将上述浓缩液转移至KD 浓缩管中,氮吹浓缩到1~2 mL,氮气流下转移至微量进样小瓶中,用正己烷洗涤KD 管,一并转入进样瓶浓缩到约100 μL。加入PCDD/Fs 回收率内标标准溶液(B.3)10 μL 和DL-PCBs 回收率内标标准溶液(B.10×100,异辛烷)40 μL,继续在微小氮气流下浓缩至约20 μL,供GC-MS/MS 测定。如果样品当日不进行仪器分析,则于-10 ℃下避光保存。
1.2.2 仪器分析条件 PCDD/Fs 测定条件:色谱柱:SHIMADZU SH-Rxi-5Sil MS(60 m×0.25 mm×0.25 μm 膜厚);柱升温程序:初始温度120 ℃,保持1.0 min,以43 ℃/min 升温至220 ℃,保持15 min,再以2.3 ℃/min 升温至250 ℃,以0.9 ℃/min升温至260 ℃,以20 ℃/min 升温至310 ℃,保持9 min;接口温度290 ℃,载气为氦气(纯度不小于99.999%),流量为1 mL/min,恒流模式;离子源:电子轰击源(EI),温度230 ℃,电离能量70 eV。进样量1 μL;灯丝电流0.5 kV;溶剂延迟时间4.5 min。
DL-PCBs 测定条件:色谱柱SHIMADZU SHRxi-5Sil MS(60 m×0.25 mm×0.25 μm 膜厚);柱升温程序:初始温度80 ℃,保持2.0 min,以15 ℃/min 升温至150 ℃,以2.5 ℃/min 升温至270 ℃,保持3 min,以15 ℃/min 升温至310 ℃,保持3 min;接口温度290 ℃,载气为氦气(纯度不小于99.999%),流量为1.2 mL/min,恒流模式;离子源:电子轰击源(EI),温度230 ℃,电离能量70 eV。进样量1 μL;灯丝电流0.5 kV;溶剂延迟时间6.0 min。
采集方式:多反应离子监测模式(MRM),母离子/子离子对及其碰撞能量见表1。
2 结果与分析
2.1 色谱柱及仪器检测条件的选择
色谱分离时,固定液种类的不同极大地影响目标化合物在色谱柱上的保留时间和分离。本试验对2,3,7,8-TCDF 异构体的分离进行了探讨,结果如下。选用了Agilent J&W HP88(100 m×0.25 mm,膜厚0.20 μm)、Agilent DB-5(50 m×0.32 mm,膜厚1.2 μm)、RTX-2330(60 m×0.25 mm,膜厚0.1 μm)3 种不同型号及规格的毛细管色谱柱,来考察TCDD/TCDFs 的分离度和时间窗口。当选用HP-88、DB-5 色谱柱时,2,3,7,8-TCDF 的分离度达不到标准要求,目标物的出峰不明显,因此都无法确定目标物的保留时间。当选用RTX-2330色谱柱时,2,3,7,8-TCDF 分离效果较好,因此进一步优化气相的方法,以达到最优的分离度。接下来对进样口温度(260,280 ℃),起始柱温(80,90,100,110,120 ℃),进样量(1,2 μL)进行了研究,结果发现,进样口温度和进样量对分离度的影响不大,而起始柱温影响2,3,7,8-TCDF 的分离,在上述设定的5 种温度下,结果发现120 ℃下,2,3,7,8-TCDF 的峰形较好,能够达到规定的分离度。
图1、2 分别为PCDD/Fs 和DL-PCBs 校正标准溶液的MRM 色谱图(CS3)。
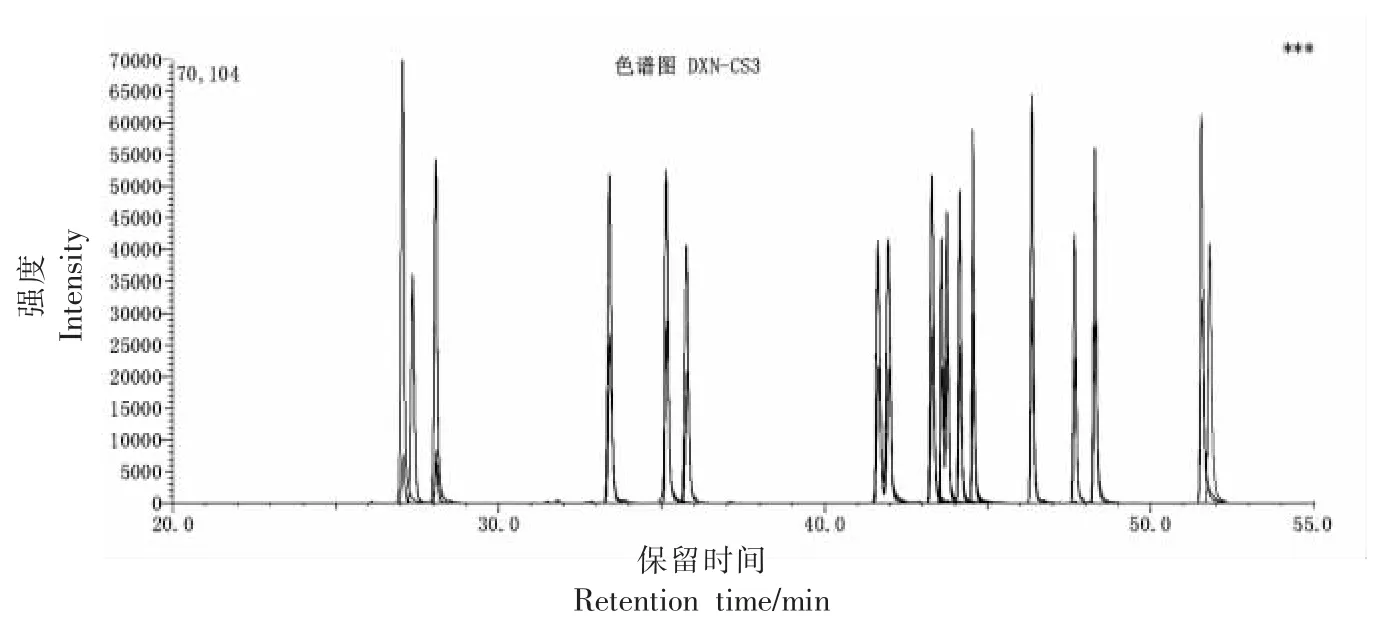
图1 PCDD/Fs 校正标准溶液的MRM 色谱图(CS3)Fig.1 Chromatograms of PCDD/Fs solvents(CS3)
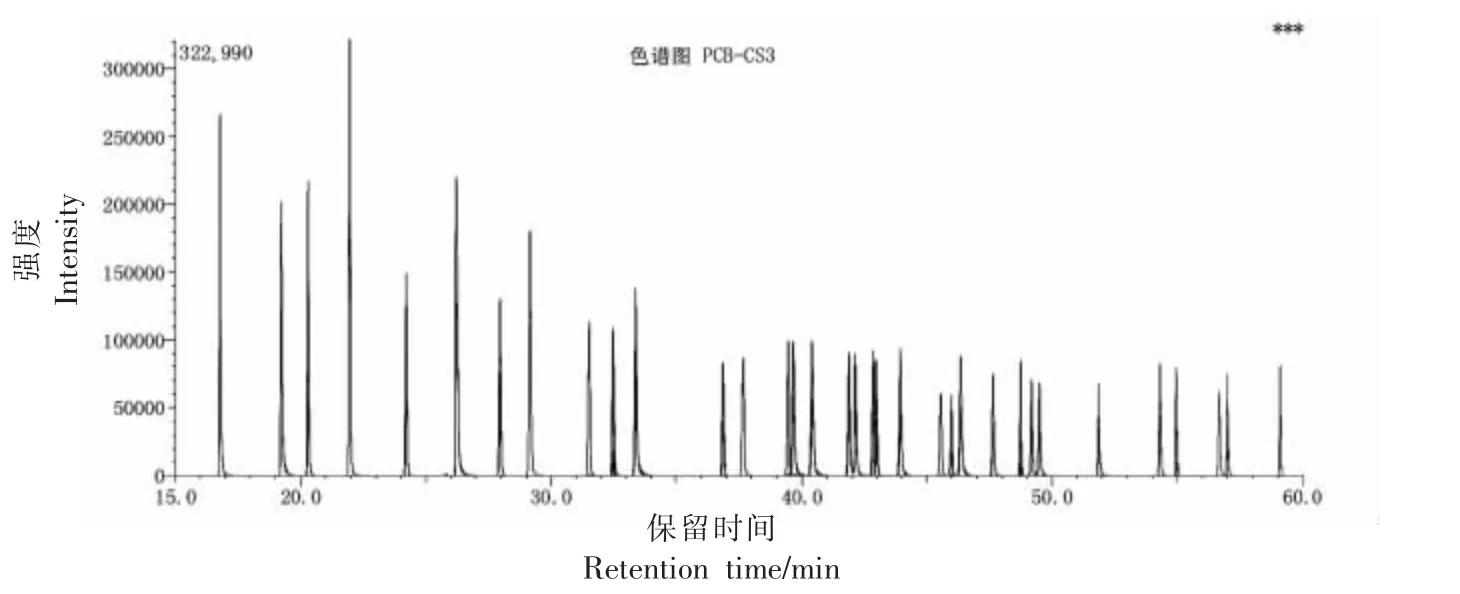
图2 DL-PCBs 校正标准溶液的MRM 色谱图(CS3)Fig.2 Chromatograms of DL-PCBs solvents(CS3)
2.2 样品前处理过程探讨
因鱼肉脂肪含量较低,且水产品为关区主要的检测样品类型,因此,首先选取鱼肉为样品基质,采取快速溶剂萃取仪进行提取,得到提取液,然后以全自动样品净化系统进行净化,在净化过程中,发现提取、净化步骤、洗脱液等主要影响因素[27-28],分析如下:
当提取循环1 次时,样品提取不完全,因此,为了增加目标物的回收率,增加了提取的循环次数和时间,这和文献报道的结论一致[29-30]。
根据标准[16],在同一批样品分析之前,应首先进行分析过程的精密度和回收率试验,样品中同位素内标的回收率需达到25%~150%。结果发现,77L 和81L 的回收率分别为16.3%,13.5%。因此接下来对这两个内标的损失来源进行了探讨。取B.8×10 溶液20 μL,加入正己烷100 mL 混匀,然后对此溶液分别只进行酸化硅胶净化、全自动净化仪净化。结果发现,经过酸化硅胶净化后77L 和81L 的回收率分别为73.1%,73.8%,经过全自动净化仪净化后77L 和81L 的回收率分别为38.4%,31.5%,由此可见,全自动净化仪对目标物的损失影响较大。接下来考察净化仪的净化步骤[30]。
净化步骤如表2 所示,多层硅胶、碱性氧化铝柱和活性炭柱经过正己烷预润洗,50%乙酸乙酯/50%甲苯、50%二氯甲烷/50%正己烷、正己烷等活化后,上样,用正己烷淋洗多层硅胶柱和铝柱后,以二氯甲烷/正己烷、乙酸乙酯/甲苯洗脱DLPCBs 后,最后活性炭柱经反冲,用甲苯洗脱PCDD/Fs。在初始净化步骤20~24 步中,系统流出溶液直接排至废液,对DL-PCBs 的回收率造成较大损失,可能跟DL-PCBs 在管路中的残留以及在乙酸乙酯/甲苯溶液中有较大溶解性有关。因此,调整洗脱步骤,20~24 步全部收集到DL-PCBs 的接收液中。同时对上样后每步的淋洗和洗脱液分别进行收集,研究77L 和81L 的损失情况,具体收集情况如下:进样前空运行一次收集液(S0)、进样后收集正己烷洗脱液(S1)、收集2%二氯甲烷/98%正己烷洗脱液(S2)、收集50%二氯甲烷/50%正己烷洗脱液(S3)、收集50%乙酸乙酯/50%甲苯洗脱液(S4)、收集正己烷洗脱液(S5)、收集甲苯洗脱液(S6)、收集再运行一次甲苯洗脱液(S7)。如图3 所示,77L 和81L 主要收集在S4 步骤中,回收率>60%。而在S5 步骤中也有少量洗脱,约为20%。在S0 和S7 步骤中只有微量的目标物出现,可忽略不计。为了进一步增加77L 和81L 的回收率,增加了50%乙酸乙酯/50%甲苯洗脱液的体积,当增加其洗脱体积由16 mL 至30 mL 时,目标物的回收率增加至80%,同时在S5 环节的残留只有1%,基本可以忽略。然而,新的问题又出现了,因为PCDD/Fs 主要集中在S6 步骤的洗脱液中,当增加S4 的洗脱体积后,最后在以甲苯洗脱PCDD/Fs时,13C12-1,2,3,4,6,7,8-HpCDF、13C12-1,2,3,4,6,7,8-HpCDD、13C12-1,2,3,4,7,8,9-HpCDF、13C12-OCDD 的回收率均小于55%,如图4 所示。前述几种目标物有部分损失,可能是在用50%乙酸乙酯/50%甲苯洗脱DL-PCBs 时,PCDD/Fs 同时也被冲到炭柱前端,从而影响PCDD/Fs 的甲苯反冲洗脱,因此,最后还是选择4 mL 的体积,而增加1次甲苯洗脱,对PCDD/Fs 的回收率影响不大,最终舍去该步骤。
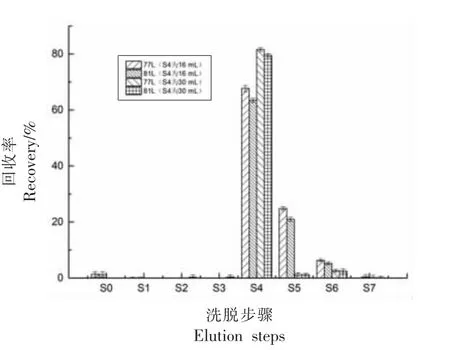
图3 DL-PCBs 在净化步骤中的洗脱情况Fig.3 Elution of DL-PCBs in purification step
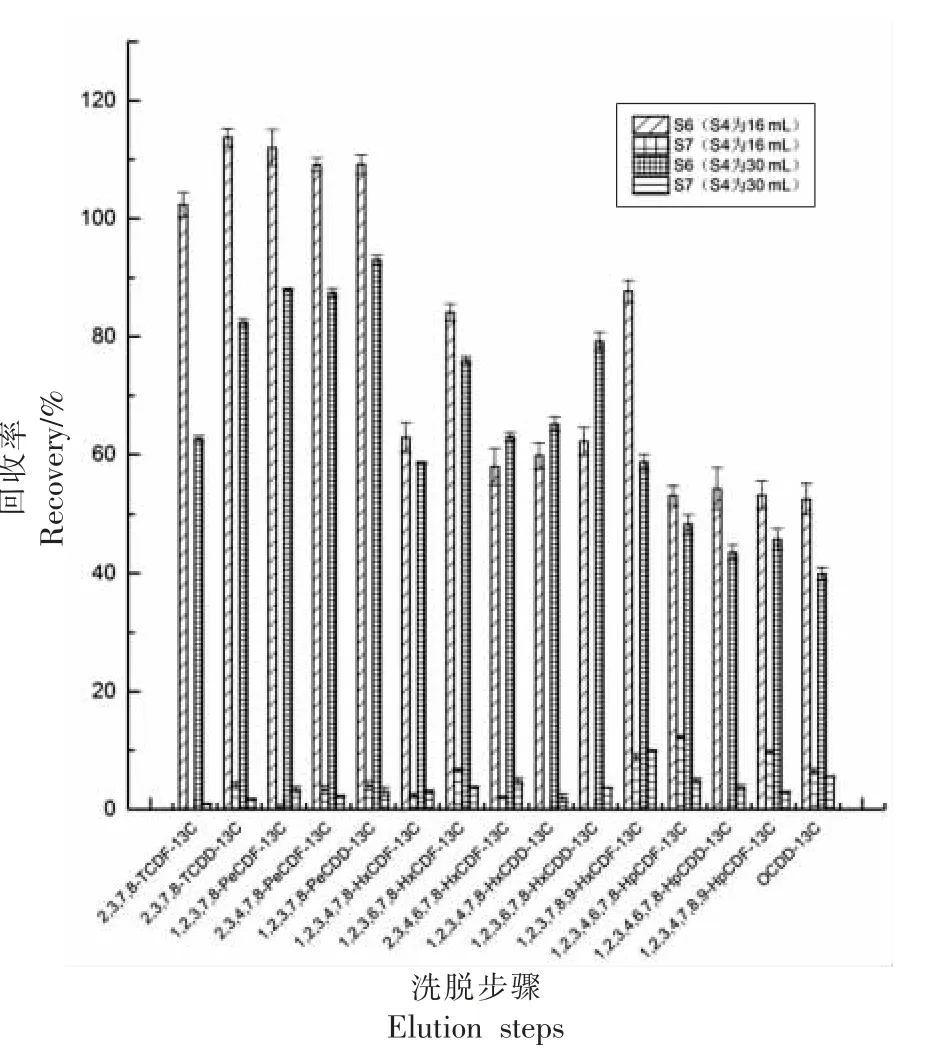
图4 PCDD/Fs 在S6 和S7 步骤中的洗脱情况Fig.4 Elution of PCDD/Fs in purification step of S6 and S7
在初始的净化步骤中,20~24 步溶液没有收集,PCDD/Fs 的测定峰形较好,当收集此部分溶液后,发现PCDD/Fs 的测定峰形变差,容易受DLPCBs 干扰。因此,在后续试验中,分开收集DLPCBs 和PCDD/Fs,分开测定。当测定完成PCDD/Fs 后,将其和DL-PCBs 的溶液合并,氮吹至约20 μL,达到完整收集和测定DL-PCBs 的目的,可以提高DL-PCBs 的回收率。77L 和81L 的回收率分别为93.0%,97.6%,满足方法要求。
在上述方法考察中发现,S0 步骤中目标物的含量可以忽略不计,这也从侧面印证了仪器基本不存在残留和交叉污染问题。但是试验发现,全自动净化仪在程序运行期间如有停电、故障等原因造成的停机、死机时,则容易导致目标物的残留,出现交叉污染情况,则需要用空白溶剂反复循环多次才能消除系统污染。因此,在仪器运行期间,需要保证仪器的正常运行。
2.3 方法的线性、检出限和回收率
将PCDD/Fs 校正标准溶液和DL-PCBs 校正标准溶液分别按浓度由低到高的顺序注入GCMS/MS 中,得到峰面积。以目标物的含量为横坐标,峰面积为纵坐标,根据内标法定量,绘制标准曲线,求出线性相关方程,各物质的线性方程见表3,线性相关系数R2>0.999,表明该方法在较宽的浓度范围内色谱峰面积与含量呈较好的线性关系。从表3 可知,仪器测定时,各化合物的相对保留时间为1.000~1.001,符合国家标准的要求。同时,表3 还列出了CS1 测定时的S/N 值,可见S/N值>3,符合检出限的计算法则,因此本方法能够达到国家标准中的限量要求[16]。
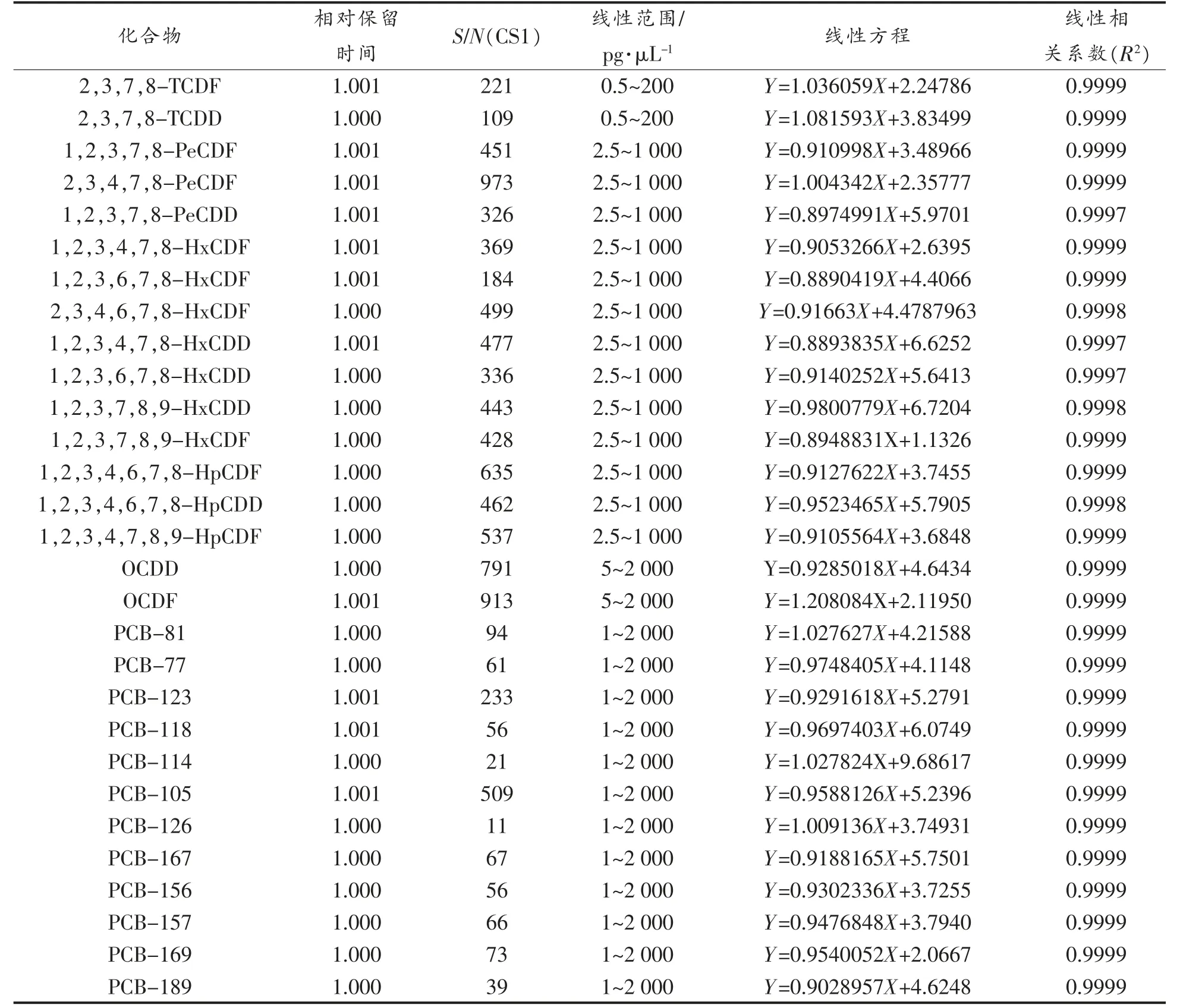
表3 PCDD/Fs 和DL-PCBs 的线性方程Table 3 The line equation of PCDD/Fs and DL-PCBs
以鱼肉为例,进行分析过程的精密度和回收率(OPR)试验,加入PCDD/Fs 和DL-PCBs 回收率和精密度标液(B.4×10 和B.11×100)各50 μL,按上述方法进行样品前处理并分析测定,采用内标法定量,设3 组平行试验,得到的内标回收率结果见表4,目标物的回收率结果见表5。内标的回收率为63.3%~106.8%,RSD 为0.4%~9.6%;目标物的回收率为93.6%~114.5%,RSD 为0.9%~9.2%。因此,该方法准确可靠,符合标准的要求,可以满足实际鱼肉样品中PCDD/Fs 和DL-PCBs 的测定要求[16]。
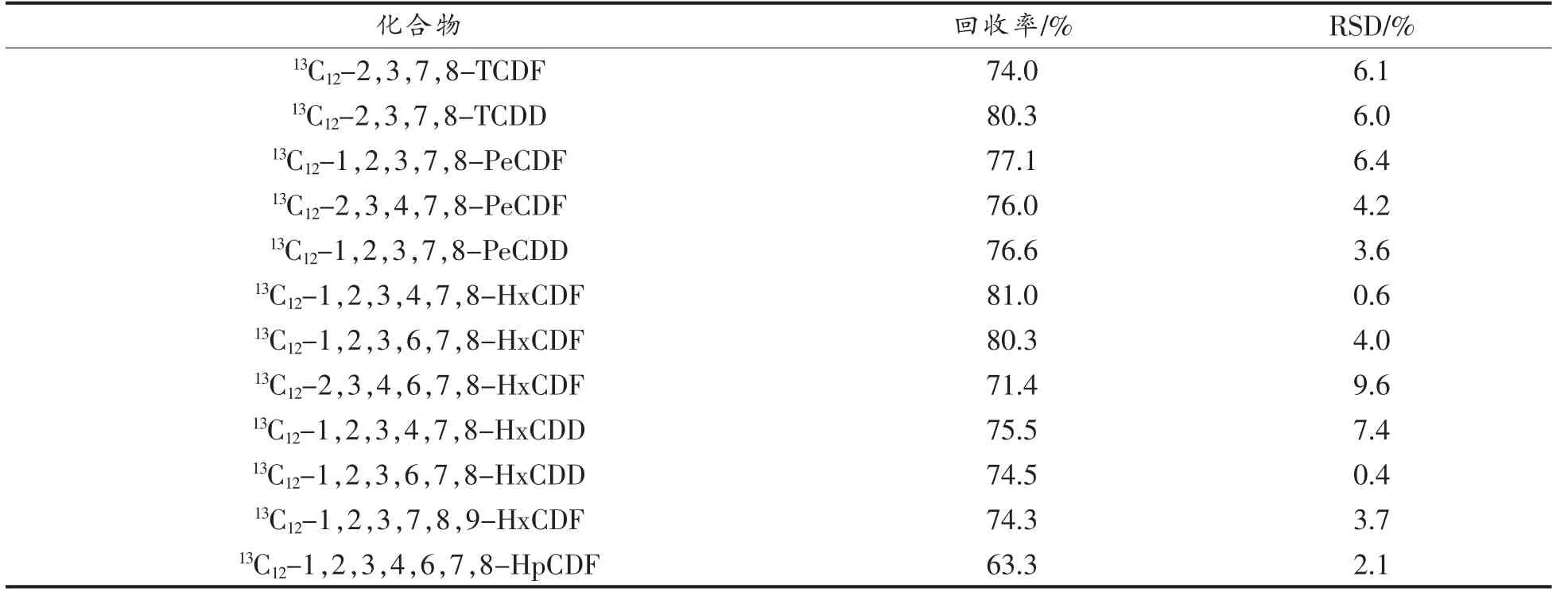
表4 鱼肉样品中PCDD/Fs 和DL-PCBs 的内标回收率和相对标准偏差(n=3)Table 4 Recoveries and relative standard deviations(RSD)of PCDD/Fs 和DL-PCBs in fish(n=3)
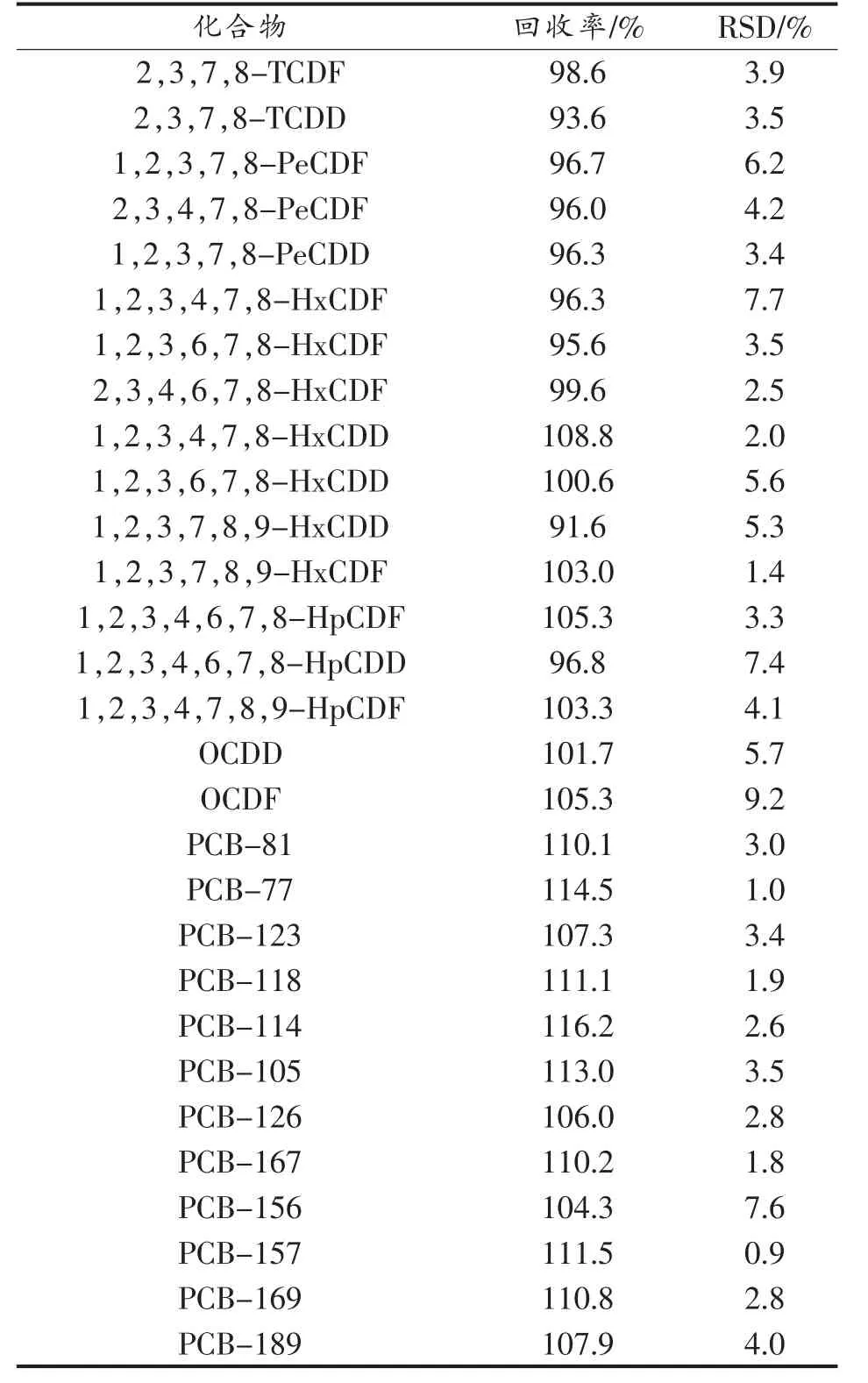
表5 鱼肉样品中PCDD/Fs 和DL-PCBs 的回收率和相对标准偏差(n=3)Table 5 Recoveries and relative standard deviations(RSD)of PCDD/Fs and DL-PCBs in fish(n=3)
3 结论
本文对采用GC-MS/MS 法测定鱼肉样品中PCDD/Fs 和DL-PCBs 的测定步骤进行了方法优化和探讨,给前处理及仪器方法提供了更为详细的解说。试验结果表明,方法回收率和精密度、各化合物的方法检出限等都符合国家标准要求,方法具有普适性,为保障食品安全提供了很好的技术支持。
