铽-磺酰基杯[4]芳烃单核配合物的单晶结构转变和发光性质
2023-11-24毛东骜徐林猛毕研峰
毛东骜, 徐林猛, 毕研峰
铽-磺酰基杯[4]芳烃单核配合物的单晶结构转变和发光性质
毛东骜, 徐林猛, 毕研峰
(辽宁石油化工大学石油化工学院, 抚顺 113001)
由对叔丁基-磺酰基杯[4]芳烃(H4TC4A-SO2)和Tb(CH3COO)3制备了一例单核铽-杯芳烃配位化合物 (Tb1-A). 杯芳烃能够敏化铽离子, 使Tb1-A无论固体状态还是在甲醇溶液中均能有效发光. 通过荧光滴定发 现, 1,10-菲啰啉(phen)与Tb1-A存在两种明显不同的作用模式, 并可影响其发光性能. 通过单晶结构分析表征了滴定过程中的两个单核化合物(Tb1-B和Tb1-C), 确定了phen与Tb1-A两种不同的作用模式, 其中一种作用模式是phen与稀土离子的配位作用, 另一种作用模式是phen与杯芳烃的主-客体相互作用. 两种作用模式对稀土发光的影响不同. 结合理论计算结果发现, 客体分子与杯芳烃间的主-客体相互作用是导致荧光变化的主要因素.
稀土; 杯芳烃; 单晶结构转变; 发光性质
镧系配合物具有丰富的能级结构和特殊的-电子跃迁, 具有发光寿命长、 发光效率高和Stokes位移大等优点, 被广泛用于发光[1~3]、 催化[4,5]以及磁性[6,7]等领域. 镧系配合物作为发光材料, 其发光效率主要取决于“天线”配体的光吸收性质以及配体-金属之间的有效能量传递(LMET). 客体分子, 包括溶剂分子, 也可以与配合物主体结构相互作用而改变化合物的基态和激发态, 从而影响配合物的发光性质[8]. 因此, 理解和调控稀土离子、 有机配体以及客体分子的相互作用机制, 是设计制备高性能稀土发光材料的关键.
杯芳烃是一类通过亚甲基或者硫原子酚类单元形成的富电子环状化合物, 因其酚单元数目可调且有多变的构象, 同时上缘(叔丁基)与下缘(酚羟基)可进行修饰功能化, 是构建多功能金属配合物的优良配体[9~13]. 叔丁基磺酰基杯[4]芳烃(H4TC4A-SO2)作为杯芳烃的一类重要衍生物, 已被广泛用于构建各种过渡金属-杯芳烃多核化合物[14~16]. H4TC4A-SO2也易与稀土离子形成单核稀土-杯芳烃(Ln-TC4A-SO2)次级结构单元(SBUs), 再通过桥连配体连接, 可形成双核[17]、 八核[18]或十二核[19]等结构的高核稀土-杯芳烃配位化合物. 近来有研究证实, H4TC4A-SO2作为“天线”配体能选择性敏化不同稀土离子发光且发光性能依赖于杯芳烃构象[17], 而客体分子与稀土-杯芳烃SBUs的相互作用又可影响化合物的发光性能, 发光机制较复杂[20]. 因此, 了解Ln-TC4A-SO2SBU的配位性质以及相互作用规律, 能为构建高效稀土-杯芳烃发光材料提供依据.
本文合成并表征了3例单核Tb-TC4A-SO2配合物{[Tb(TC4A-SO2)(CH3OH)2(H2O)2][Tb(TC4A-SO2)·(CH3OH)(H2O)2](TBA)2}(CH3OH)2(H2O)4(Tb1-A), {[Tb(TC4A-SO2)(phen)(CH3OH)2](TBA)}·(CHCl3)2(Tb1-B)和{[Tb(TC4A-SO2)(phen)(H2O)2](TBA)}(phen)2(CH3OH)2(Tb1-C)(phen=1,10-菲啰啉; TBA=四丁基铵根离子), 讨论了溶剂分子和辅助配体(配位和客体)对配合物发光性质的影响, 并通过荧光滴定与DFT计算等方法分析了单晶结构转变与发光性质变化之间的关系.
1 实验部分
1.1 试剂与仪器
参照文献[21]方法合成H4TC4A-SO2. 六水合醋酸铽[Tb(CH3COO)3·6H2O), 纯度99.99%]和1,10-菲啰啉(phen), 阿拉丁生化股份有限公司; 四丁基溴化铵(TBAB), 分析纯, 麦克林生化科技有限公司; 无水甲醇和三氯甲烷, 分析纯, 国药集团化学试剂有限公司.
Bruker D8 QUEST型单晶X射线衍射仪, 德国Bruker公司; TA Q600型热重-差热分析仪(TGA-DSC), 美国TA公司; FTIR-660I610 型红外光谱仪, 美国安捷伦科技有限公司; F-4500 FL型荧光光谱仪, 日本日立高新技术公司.
1.2 实验过程
1.2.1化合物Tb1-A/B/C的合成将Tb(CH3COO)3·6H2O(0.036 g, 0.1 mmol)、 H4TC4A-SO2(0.085 g, 0.1 mmol)、 TBAB(0.16 g, 0.48 mmol)、 水(70 μL)、 无水甲醇(10 mL)和三氯甲烷(10 mL)混合, 反应3 h后过滤, 滤液静置培养至有大量无色方块状晶体析出, 即为Tb1-A.
上述相同条件下, 加入phen(0.04 g, 0.2 mmol), 可得到无色菱形晶体Tb1-B; 过滤晶体, 将母液继续静置, 析出无色片状晶体Tb1-C. 晶体图见图 S1(见本文支持信息). 所得晶体于80 ℃真空干燥后, 计算Tb1-A/B/C的产率分别为80%, 60%和10%(基于H4TC4A-SO2).
1.2.2单晶结构表征通过Bruker D8 QUEST型单晶X射线衍射仪(Mo射线,= 0.071073 nm)收集化合物的单晶衍射强度数据. 利用F全矩阵最小二乘法还原数据, 并通过SHELXTL⁃2014程序进行解析[22]. 氢原子采用理论加氢, 未加氢原子直接加入分子参与精修. 除氢原子外, 其它原子采用各向异性修正. 化合物Tb1-A/B/C的晶体学数据见表1. 精修数据已存于剑桥晶体学数据库(CCDC: 2238005~2238007).

Table 1 Crystal data and structure refinement for Tb1-A/B/C
1=Σ||0|-|c||/Σ|0|;2={Σ[(02-c2)2]/Σ[(02)2]}1/2.
1.3 密度泛函理论计算
密度泛函理论计算采用Gaussian 16软件包, 并通过化合物单晶数据建立模型进行计算[23,24]. 密度泛函理论中的杂化泛函(B3LYP)方法采取6⁃31G(,)基组. 因分子整体结构较大, 原子数较多且变量复杂, 无法实现分子整体结构有效计算, 仅以3例化合物中的对叔丁基磺酰基杯[4]芳烃配体和邻菲罗啉配体的基态构型进行限制性结构优化计算. 优化后的结构采取同样的基组与方法计算最高占据轨道(HOMO)和最低未占据轨道(LUMO)的能量.
2 结果与讨论
2.1 Tb1-A/B/C的合成和单晶结构
通常, 共轭型配位分子与稀土离子结合会增加分子的刚性, 减少非辐射跃迁, 有利于配体到稀土离子的电子跃迁, 从而提升稀土离子的发光性能[20,25]. 但在研究Tb-TC4A-SO2发光体系时发现1,10-菲啰啉(phen)所起的作用恰恰相反. 在图1所示的体系中, 伴随着phen配体浓度的变化, 通过重结晶方式得到了3种不同的化合物晶体(Tb1-A/B/C). 通过单晶X射线衍射表征了化合物的结构, 并结合荧光滴定实验, 从原子分子层面研究了phen对Tb-TC4A-SO2发光体系的影响.
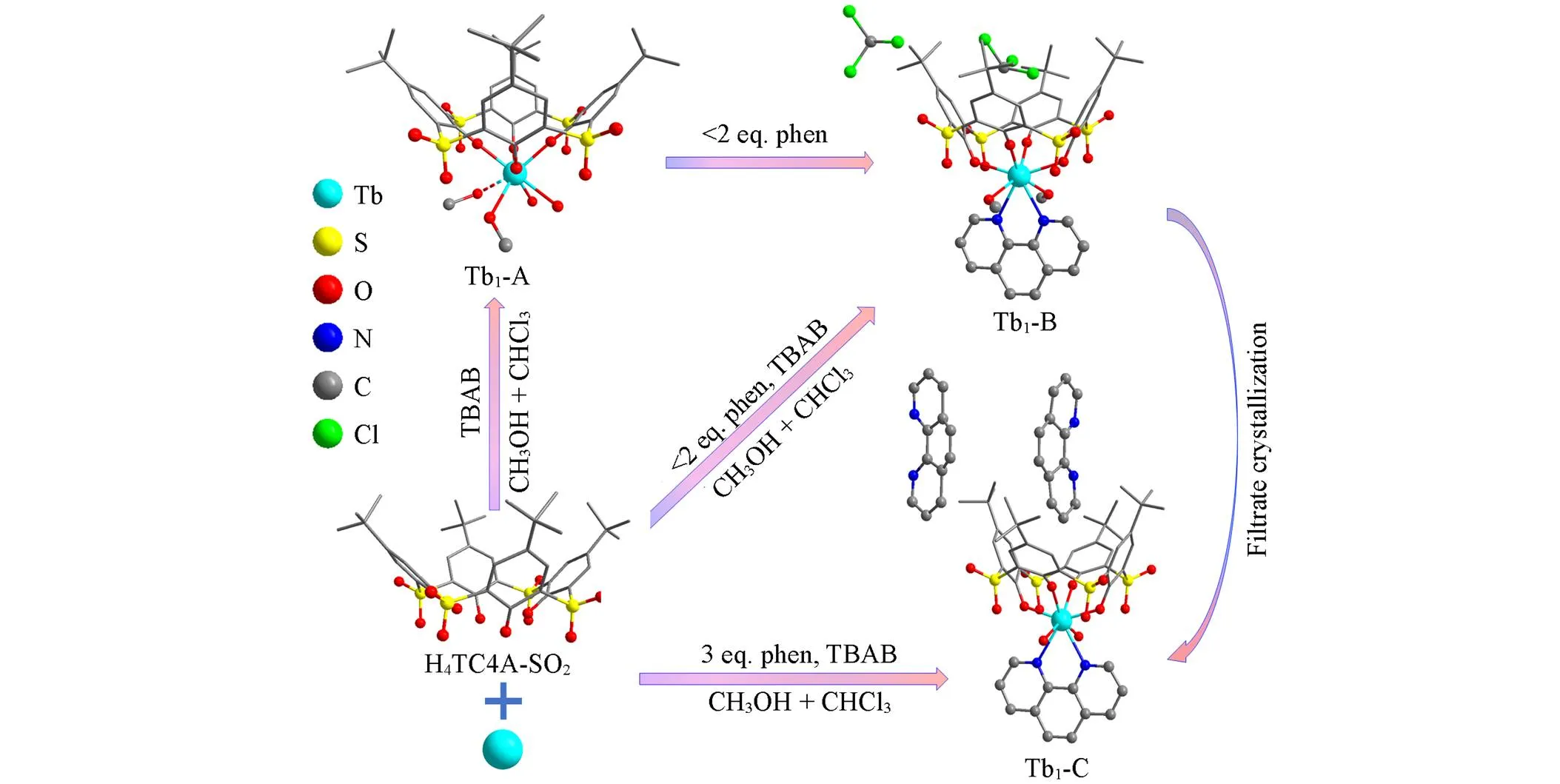
Fig.1 Crystal structure transition of Tb1⁃TC4A⁃SO2 complexes(Tb1⁃A/B/C)
稀土铽离子与H4TC4A-SO2在摩尔比1∶1的条件下即可结晶得到Tb1-A. 单晶结构解析发现, Tb1-A属于单斜晶系, 空间群为. 每个解析不对称单元中含有2个TbⅢ稀土中心和2个TC4A-SO2配体. 2个TbⅢ均与杯芳烃配体的4个酚羟基氧配位, 其中1个TbⅢ还与2个甲醇分子和2个水分子配位, 形成八配位环境(图S2, 见本文支持信息); 另外1个TbⅢ与2个水分子和1个甲醇分子形成七配位模式.
向Tb1-A或者Tb1-A合成体系中加入phen, 且浓度低于TbⅢ的2倍时会生成Tb1-B. Tb1-B结晶为三斜晶系, 空间群为Tb1-B中八配位TbⅢ中心与杯芳烃的4个酚羟基配位点的结合模式与Tb1-A一致. 区别是螯合配位的phen占据了配位点, 2个甲醇分子与金属中心配位, 位于phen的两侧. 这说明从Tb1-A到Tb1-B的过程中, 除杯芳烃的配位点外, 其余配位点因为phen的配位作用实现了重排. 需要特别指出的是, 在Tb1-B的结构中, 杯芳烃的杯腔被1个三氯甲烷溶剂分子占据, 能够被C—H…共轭效应稳定而存在[26].
当phen浓度达到TbⅢ的3倍或在结晶分离出Tb1-B后的滤液中(phen浓度相对变大)可以结晶得到Tb1-C. 与Tb1-B结构相比, 金属离子与杯芳烃和配位的phen的结合模式不变, 区别是杯芳烃杯腔内的三氯甲烷分子被无序的phen分子取代. 高浓度的phen并未能再与稀土离子配位, 而是与溶剂分子竞争占据杯芳烃杯腔位置.
在Tb1-A/B/C中, 每个‒1价稀土阴离子TbⅢ-TC4A-SO2单元与1个四丁基铵阳离子(TBA)平衡电荷, 保持配合物整体电中性.
H4TC4A-SO2具有一个大而构象可变的的共轭体系, 电子离域在整个配体上, 与稀土离子形成配合物的发光性质受杯芳烃配体构象结构的影响[18]. 从Tb1-A到Tb1-C, 随着phen的配位以及杯芳烃的包合, Tb1-A结构中的2个配位溶剂分子被phen所取代, 同时与phen平面平行的2个Tb—O酚键距离不断减小, 位于phen平面两侧的2个Tb—O酚键距离不断增大(键长和键角信息见本文支持信息表S1和 表S2). 杯芳烃中对位叔丁基碳之间的距离从1.1676 nm(1.0433 nm)变化到1.264 nm(0.8996 nm), 同时2个对叔丁基酚单元之间的二面角从87.66°(141.71°)变化到67.31°(148.44°) . 以上结果均证明, 在Tb1-A → Tb1-B → Tb1-C过程中, 杯芳烃的杯腔变形程度逐渐变大(图2).
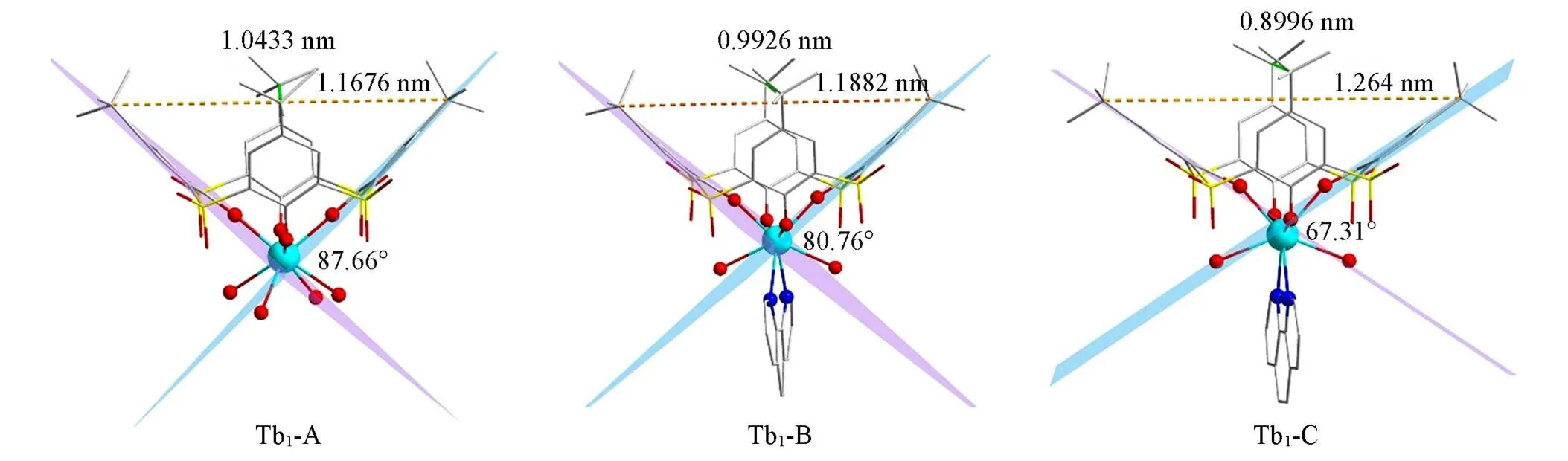
Fig.2 Structures of SBU⁃A/B/C and conformation changes of TC4A⁃SO2
化合物Tb1-A的堆积结构中单核SBU(SBU-A)呈“头斜对着头” 的排列方式(图3), 形成分子胶囊状结构, 能够容纳多个客体分子形成结晶微环境[27,28]. 因此该结构容易被某些溶剂分子所分散. 与Tb1-A相比, Tb1-B中多了1个与金属中心配位的phen, 杯芳烃仍然采取“头斜对着头”的排列方式. 但在 Tb1-B 中, 每2个稀土-杯芳烃单元(SBU-B)中的杯腔可以分别容纳1个三氯甲烷, 形成包合2个客体分子的胶囊结构. 化合物Tb1-C的阴离子单元结构单元(SBU-C)与Tb1-B的类似, 只是2个SBU-C胶囊中封装了phen分子而非三氯甲烷分子. phen分子与杯芳烃之间以C—H…和…作用相互连接[29]. SBU-C 排列方式与SBU-A和SBU-B不同, 呈现 “头正对头”的排列方式. Tb1-A/B/C中, 稀土-杯芳烃单元SBU与客体分子不同作用所形成的胶囊状结构中, 金属离子之间的距离也从1.31 nm(SBU-A),到1.62 nm(SBU-B), 最终达到1.71 nm(SBU-C)(图S3, 见本文支持信息).
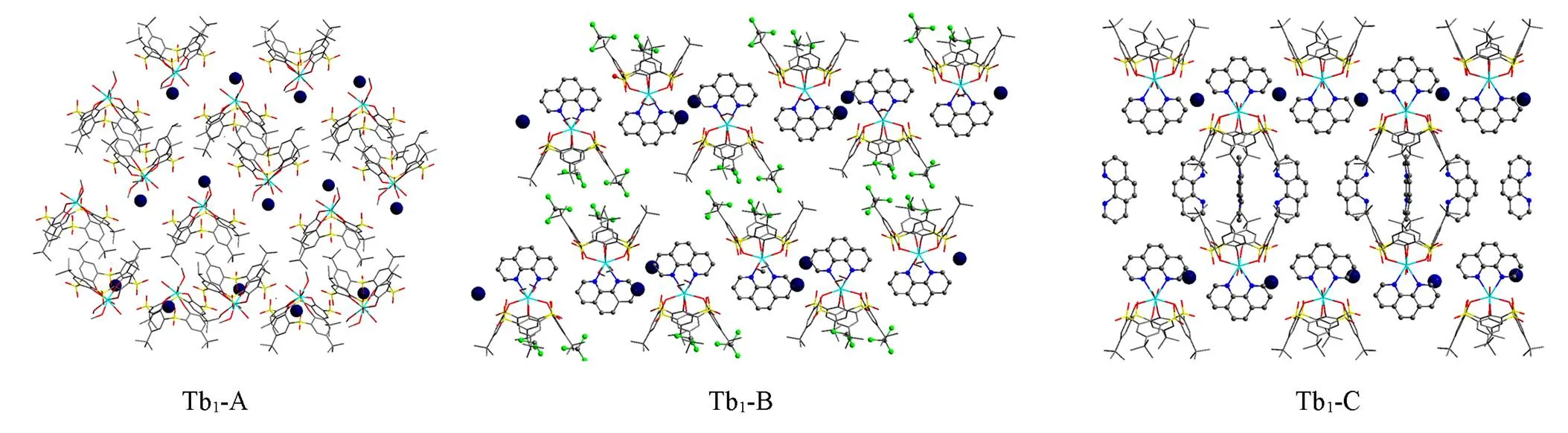
Fig.3 Packing diagrams of Tb1⁃A/B/C
The blue ball represents TBA.
另外, 在红外图谱中发现了Tb1-A/B/C有机官能团的特征峰. 通过热重实验研究了3个化合物的热稳定性, 具体实验结果和分析见本文支持信息图S4和S5.
2.2 化合物Tb1-A的溶液荧光性质
H4TC4A-SO2和Tb以1∶1的摩尔比形成的化合物具有较好的发光性能. 在这一比例条件下结晶得到的Tb1-A化合物在甲醇中有较好的溶解性和良好的发光性能. 用phen对Tb1-A的甲醇溶液进行荧光滴定, 测试结果如图4(A)所示. 加入phen后, Tb1-A的发光强度呈减弱趋势. 滴定结果可根据Stern-Volmer方程0=1+[1][Q](用于静态线性猝灭)和0=e2[Q]+B(用于指数猝灭)来更好地理解发光强度降低的机制(式中,0为在未添加phen的情况下, 纯Tb1-A的荧光强度;为phen存在下的荧光强度; [Q]为分析物的摩尔浓度;1或2为SV常数, 量化猝灭效率; A和B为常数)[13]. phen对Tb1-A荧光强度的影响可由两段拟合关系表示[图4(B)], phen浓度范围在0~2 mmol/L之间采取静态线性猝灭, 线性 方程为00.073[phen]+0.994(2=0.972); 当phen浓度大于2 mmol/L时采取指数猝灭, 线性方程为0/=1.066e0.127[phen]‒0.201(2=0.999). 在phen滴定Tb1-A过程中, 体系的发光性能与晶态化合物 Tb1-A→Tb1-B→Tb1-C在甲醇中的发光减弱的趋势一致[图4(C)]. 对于Tb1-A而言, 当[phen]∶[Tb1-A]达到1∶1之前, Tb1-A可与phen结合生成Tb1-B, phen与金属离子配位; 当[phen]∶[Tb1-A]大于1∶1之后, phen不仅与金属离子配位, 还进入到TC4A-SO2配体的杯腔, 生成Tb1-C. 因此, 具有线性关系的猝灭过程是由phen的配位导致, 而具有动态相互作用的发光减弱是由phen与杯芳烃杯腔的相互作用导致[30]. 将晶体溶于甲醇中测试了荧光发射(相同Tb浓度)光谱, 发现3个单核配合物发光性质的趋势与溶液中的趋势一致, 即以Tb1-A→Tb1-B→Tb1-C顺序逐步减弱[图4(C)].
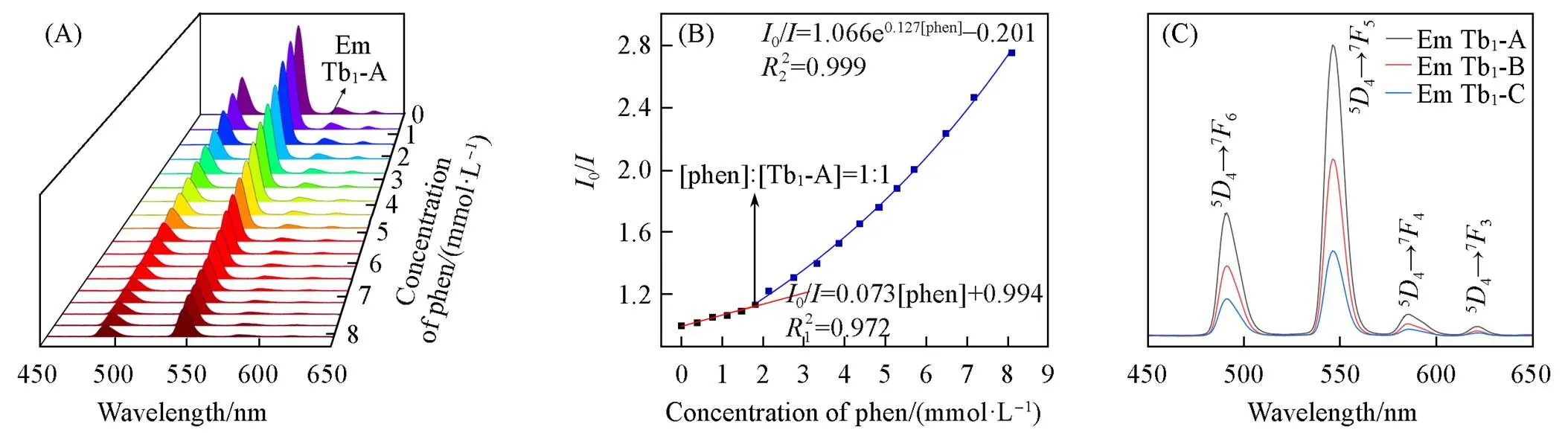
Fig.4 Luminescence of Tb1⁃A after addition of phen
(A) Emission spectra; (B) Stern-Volmer plots; (C) emission spectra of Tb1methanol solution(ex=388 nm).
2.3 DFT计算
配合物的前线分子轨道, 特别是最高占据分子轨道(HOMO)和最低未占据分子轨道(LUMO)对配合物的光学性质有着较大影响. 以单晶结构数据为基础搭建计算模型, 对Tb1-A/B/C中配位的杯芳烃以及Tb1-B/C配位的phen的轨道能级进行了量化计算. 计算结果(图5)显示, 从化合物Tb1-A到Tb1-C, 杯芳烃的能隙逐渐增高, 说明与铽的匹配程度降低, 配合物发光性能减弱. 配位phen的HOMO、 LUMO能量与能隙均高于Tb1-A/B/C中配位的杯芳烃. 因此在给定的激发波长下(=388 nm), 无法将phen激发到稀土离子的激发态, 而此时杯芳烃到稀土离子可有效激发, 会将能量传递到phen. 由于荧光光谱上并未见到phen的发射峰, 因此稀土传递到phen的能量会传递回稀土离子. 当能量从phen再返回到稀土铽中心时存在能量损失, 如非辐射跃迁等, 造成荧光减弱[3]. 综上所述, Tb1-A → Tb1-B → Tb1-C化合物发光减弱的原因是客体分子(包括溶剂)与杯芳烃的主-客体作用改变了TC4A-SO2的构象, 从而减小了配体-金属能量传递(LMET)效率, 最终影响了配合物的发光.
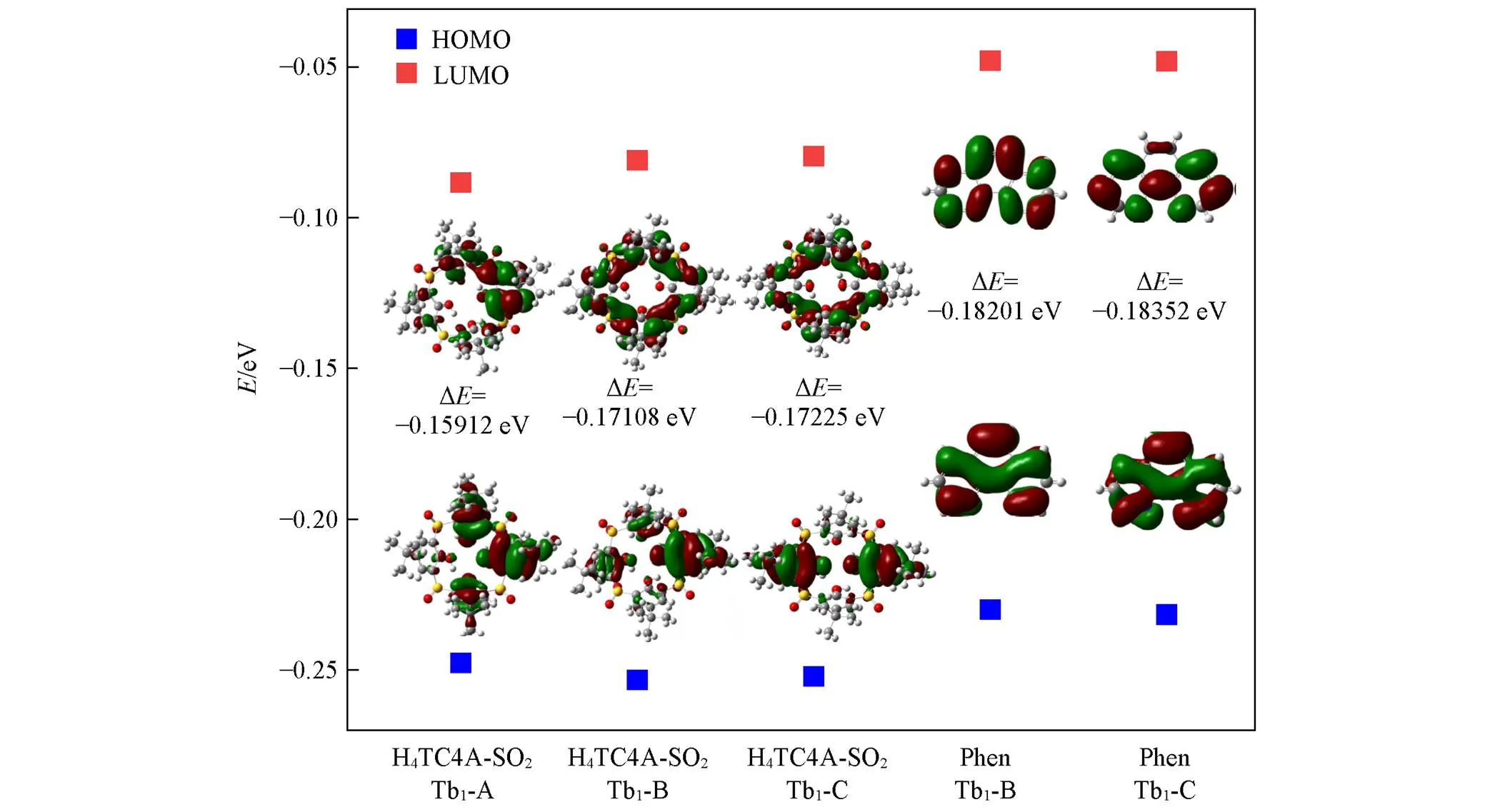
Fig.5 Molecular orbitals and energy gaps of the ligands
3 结 论
在Tb-TC4A-SO2发光体系中, 进行了客体分子phen荧光滴定实验, 得到了3个化合物的单晶结构, 并进行了相关的理论计算. 实验结果表明, 客体phen使Tb-TC4A-SO2主体化合物发光变化是由客体与主体化合物两种不同作用模式导致的. phen与稀土离子的配位模式会导致能量的辐射跃迁, phen与Tb-TC4A-SO2中杯芳烃的杯腔作用会改变杯芳烃的构象, 均能通过稀土离子与杯芳烃的能级匹配差异而导致发光性能的变化. 本研究为通过主-客体相互作用调控稀土离子发光来制备高性能发光材料提供了实验基础和理论依据.
支持信息见http://www.cjcu.jlu.edu.cn/CN/10.7503/cjcu20230107.
[1] Li X. Z., Tian C. B., Sun Q. F.,, 2022,(6), 6374—6458
[2] Sahoo S., Mondal S., Sarma D.,, 2022,214707
[3] Zhang R. Y., Zhu L. L., Yue B. B.,, 2023,(2) , 214707
[4] Bell D. J., Natrajan L. S., Riddell I. A.,, 2022,108009
[5] Zhu S., Xu W., Hong D., Wu W., Chai F., Zhu X., Zhou S., Wang S.,, 2023,(1), 381—391
[6] Chen J., Carpenter S. H., Fetrow T. V., Mengell J., Kirk M. L., Tondreau A. M.,, 2022,(46), 18466—18475
[7] Wilharm R. K., Ramakrishnam Raju M. V., Hoefler J. C., Platas⁃Iglesias C., Pierre V. C.,, 2022,(9), 4130—4142
[8] Zhang X. J., Wang W. J., Hu Z. J., Wang G. N., Uvdala K.,, 2015,206—235
[9] Iki N., Kabuto C., Fukushima T., Kumagai H., Takeya H., Miyanari S., Miyashi T., Miyano S.,, 2000,(11), 1437—1443
[10] Hang X. X., Bi Y. F.,, 2021,, 3749—3758
[11] Takashi K., Nobuhiko I., Masahiro Y.,, 2007,(13/14), 1734—1746
[12] Menon S. K., Sewani M.,., 2006,(1), 49—82
[13] Li Z. P., Wang D., Zhou Z. H., Zhao G. Y., Li Q., Bi Y. F., Zheng Z. P.,, 2022,(51), 20814—20823
[14] Zhang M., Chen M. W., Gao H., Bi Y. F.,, 2019,(10), 2052—2058(张敏, 陈梦伟, 高虹, 毕研峰. 高等学校化学学报, 2019,(10), 2052—2058)
[15] Shi C., Zhang M., Hang X. X., Bi Y. F., Huang L. L., Zhou K., Xu Z. H., Zheng Z. P.,, 2018,(30), 14448—14454
[16] Shi C., Chen M. W., Han X., Bi Y. F., Huang L. L., Zhou K., Zheng Z. P.,, 2018,(6), 1329—1335
[17] Kajiwara T., Katagiri K., Hasegawa M., Ishii A., Ferbinteanu M., Takaishi S., Ito T., Yamashita M., Iki N.,, 2006,(13), 4880—4882
[18] Kajiwara T., Wu H., Ito T., Iki N., Miyano S.,2004,(14), 1832—1835
[19] Kajiwara T., Katagiri K., Takaishi S., Yamashita M., Iki N.,, 2006,(3), 349—351
[20] Jin J., Xue J. J., Liu Y. C., Yang G. P., Wang Y. Y.,, 2021,(6), 1950—1972
[21] Morohashi N., Iki N., Sugawara A., Miyano S.,, 2001,,5557—5563
[22] Sheldrick G. M.,, 2015,(1), 3—8
[23] Frisch M. J., Trucks G. W., Schlegel H. B., Scuseria G. E., Robb M. A., Cheeseman J. R., Montgomery J. A. Jr., Vreven T. K., Kudin N., Burant J. C., Millam J. M., Iyengar S. S., Tomasi J., Barone V., Mennucci B., Cossi M., Scalmani G., Rega N., Petersson G. A., Nakatsuji H., Hada M., Ehara M., Toyota K., Fukuda R., Hasegawa J., Ishida M., Nakajima T., Honda Y., Kitao O., Nakai H., Klene M., Li X., Knox J. E., Hratchian H. P., Cross J. B., Adamo C., Jaramillo J., Gomperts R., Stratmann R. E., Yazyev O., Austin A. J., Cammi R., Pomelli C., Ochterski J. W., Ayala P. Y., Morokuma K., Voth G. A., Salvador P., Dannenberg J. J., Zakrzewski V. G., Dapprich S., Daniels A. D., Strain M.C., Farkas O., Malick D. K., Rabuck A. D., Raghavachari K., Foresman J. B., Ortiz J. V., Cui Q., Baboul A. G., Clifford S., Cioslowski J., Stefanov B. B., Liu G., Liashenko A., Piskorz P., Komaromi I., Martin R. L., Fox D. J., Keith T., Al⁃Laham M. A., Peng C. Y., Nanayakkara A., Challacombe M., Gill P. M. W., Johnson B., Chen W., Wong M. W., Gonzalez C., Pople J. A.,, Gaussian Inc., Wallingford CT, 2016
[24] Gagliardi L., Truhlar D. G., Manni G. L., Carlson R. K., Hoyer C. E., Bao J. L.,, 2017,(1), 66—73
[25] Fard Z. H., Kalinovskyy Y., Spasyuk D. M., Blight B. A., Shimizu G. K. H.,, 2016,(87), 12865—12868
[26] Dielemann C., Matt D., Jones P. G., Thonnessen H.,, 2003,(Pt 5), 247—249
[27] Hardie M. J., Raston C. L.,, 2000,, 2483—2492
[28] Zheng G. L., Li Y. Y., Deng R. P., Song S. Y., Zhang H. J.,, 2008,(6), 658—660
[29] Leśniewska B., Coleman A. W., Perret F., Suwińska K.,, 2019,(3), 1695—1708
[30] Sheng T. P., Fan X. X., Zheng G. Z., Dai F. R., Chen Z. N.,, 2020,(11), 2656—2666
Structural Transformation and Luminescence Properties of Terbium-sulfonylcalix[4]arene Mononuclear Complexes
MAODongao, XULinmeng, BIYanfeng*
(,,113001,)
A mononuclear terbium coordination compound(Tb1-A) was prepared from--butyl-sulfonylcalixarene(H4TC4A-SO2) and Tb(CH3COO)3. Calixarene can effectively sensitize terbium ions, enabling luminescence of Tb1-A in both solid state and methanol solution. It was found that 1,10-phenanthroline(phen) could influence the luminescence of Tb1-A by two interaction modes in fluorescence titration. Two different contacting modes between phen and Tb1-A were also determined by the single crystal structural characterization of two mononuclear compounds(Tb1-B and Tb1-C), one of which was the coordination between phen and Tb ions, the other was the host-guest interaction between phen and calixarene. Combined with the results of theoretical calculation, it was also found that the host-guest interaction between guest molecules and calixarene was the main factor that caused the fluorescence quenching.
Rare earth; Calixarene; Crystal structure transition; Luminescence property
2023-03-14
毕研峰, 男, 博士, 教授, 主要从事配位化学研究. E-mail: biyanfeng@lnpu.edu.cn
辽宁省大学生创新创业训练计划项目(批准号: S202110148002)和国家自然科学基金(批准号: 91961110)资助.
O614
A
10.7503/cjcu20230107
2023-04-14.
Supported by the Liaoning College Student Innovation and Entrepreneurship Training Program, China(No.S202110148002) and the National Natural Science Foundation of China(No.91961110).
(Ed.: N, K, M)