利用CRISPR/Cas9系统研究REVOLUTA参与烟草叶芽发育的调控
2023-11-23王兵赵会纳余婧陈杰骆梅雷波
王兵 赵会纳 余婧 陈杰 骆梅 雷波
(1. 贵州省烟草科学研究院 烟草行业分子遗传重点实验室,贵阳 550081;2. 贵州医科大学生物与工程学院(健康医药现代产业学院),贵阳 550025)
植物叶芽由茎尖分生组织及侧生分生组织、叶原基等组成,是直接影响作物结实和产量的重要农艺性状之一,生产实践中,人们通常通过调控叶芽生长发育以达到增产增效的目的[1-2]。栽培烟草(Nicotiana tabacum)作为特种叶用经济作物之一,生产过程中因“打顶”后腋芽数量激增严重影响烟叶品质和产量。传统消除腋芽主要通过“抹杈”及化学抑芽剂处理,不仅成本高,而且易导致烟草病虫害滋生,严重制约烟叶生产高质量发展。随着CRISPR/Cas9技术在拟南芥(Arabidopsis thaliana)、水稻(Oryza sativa)、油菜(Brassica napus)等植物株型改良中的广泛应用,利用该技术调控烟草叶芽发育将会逐步实现[3-6]。当前关于烟草叶芽分子调控研究还较少,挖掘调控叶芽发育的相关基因,对于在烟草生产中减工降本和减少农药使用具有重要的意义。
HD‑Zip III(homeodomain‑leucine zipper protein III)作为植物特有的转录因子家族,广泛参与调控植物多种细胞分化,如分生组织形成、胚胎形态建成、维管组织发育、侧生器官发生及极性建立等[5,7-10]。拟南芥HD‑Zip III基因家族包括5个成员(PHV/PHB/ATHB8/REV/CNA),它们存在高度保守结构域,功能呈现冗余性,除AtHB8外,其余HD‑Zip III家族基因在叶原基、侧根原基、顶端分生组织均有不同程度的表达,而只有rev突变体表现出分枝数量显著减少,同时叶片发育异常[11-13]。此外,rev隐性纯合突变体ifl1还观察到纤维细胞的细胞壁增厚过程受阻[14]。相较于拟南芥显性突变体rev‑10d,phv/phb/rev三重基因突变体表现出相对较少的侧根数量,说明在根发育过程中PHV、PHB、REV存在协同作用[15]。拟南芥athb8/cna/rev三重基因突变体表现出比rev单突变体更多的侧枝数量, 说明在侧枝形成过程中AtHB8、CNA、REV存在拮抗作用[7]。Shi等[16]通过体内外实验证实拟南芥HD‑Zip III 转录因子家族的REV能直接结合 STM基因,使STM表达量上调,从而促使AM(axillary meristem)起始。此外,研究还发现 STM位点在不同组织中的组蛋白甲基化修饰差异会影响 REV 对 STM的表达调控。研究表明DRN/DRNL‑REV和ZPR‑REV蛋白互作调控模型影响AM起始,ZPR(LITTLE ZIPPER3)在叶腋中表达并干扰 DRN/DRNL‑REV相互作用以负调节STM表达控制AM 启动[17]。在AM起始的早期阶段ARGONAUTE10(AGO10)可能通过结合miR165/166并导致miRNA的降解,而miR165/166靶基因REV活性激活,从而实现REV表达对 AM 启动的时间和空间精确调控[18]。综上可知,HD‑Zip III转录因子成员间以拮抗或者协同方式参与调控植株的形态发育过程。
陈浣等[19]对烟草NtHD‑Zip III基因家族进行鉴定及表达数据聚类分析,结果表明相较于拟南芥HD‑Zip III基因家族数量,NtHD‑Zip III基因家族数量显著增加,共鉴定到19个,其中REV基因对于叶芽的发育是必需的。REV基因参与调节分生组织形成,但因烟草缺少rev 突变体,对REV基因在叶芽发育过程中的分子机制仍有待研究。本研究将通过CRISPR/Cas9编辑系统定向突变NtREV基因的不同单靶点,通过PCR测序鉴定NtREV基因编辑形式,以期获得烟草rev纯合突变体,探究NtREV基因不同靶点在调控烟草叶芽发育中功能,为阐述NtREV在调控烟草叶芽发育的分子机理奠定基础。
1 材料与方法
1.1 材料
试验材料为烤烟品种 K326(Nicotiana tabacum CV. K326)、农杆菌感受态细胞菌株LB4404(Agrobacterium tumefaciens)、大肠杆菌感受态细胞Top10(Escherichia coli),均由实验室提供。质粒小提取试剂盒、DNA纯化试剂盒、琼脂糖凝胶回收试剂盒、DNA Taq酶、DNA Marker等试剂均从天根生化科技(北京)有限公司购置。限制性内切酶Bsa I‑HF、Nhe I、Spe I和T4 DNA连接酶(T4 DNA ligase)购自NEB(北京)有限公司。抗生素:氨苄青霉素(ampicillin, Amp)、卡那霉素(kanamycin,Kan)、潮霉素(hygromycin, Hyg)和利福平(rifampicin,Rif)购自天根生化科技(北京)有限公司。引物合成和基因测序由上海生工生物技术有限公司完成。pYAO‑based CRISPR/Cas9植物编辑载体系统由中科院遗传与发育生物学研究所焦雨铃教授馈赠,该系统由sgRNA(single guide RNA)表达盒中间载体AtU6‑26‑sgRNA‑SK和转化植物用的终载体pCAMBIA1300‑pYAO‑Cas9两部分构成。
1.2 方法
1.2.1 靶点区域选择和pYAO‑based CRISPR/Cas9载体构建 通过数据库(Nitab v4.5: https://solgenom‑ics.net/organic/Nicotiana_tabacum/genome)检索烟草REV基因并预测REV基因外显子和内含子数量,利用(https://www.ncbi.nlm.nih.gov/cdd/?term)分析REV氨基酸序列保守结构域。利用CRISPR‑P软件(http://crispr.hzau.edu.cn)[20]在烟草REV基因目标区域搜寻靶点,通过blast 检索最佳靶点,由于编辑系统AtU6‑26‑sgRNA‑SK载体的AtU6‑26启动子与gRNA序列中间包括两个Bsa I和一个EcoR I酶切位点,使用Bsa I酶切后,会分别留下与ATTG和AAAC匹配的黏性末端,因此要求在靶标序列两端分别添加5′‑ATTG‑3′和5′‑AAAC‑3′接头引物,用黑色加粗字体表示接头引物靶位点序列为5′‑ATTGGCTCGATATTCGACAGAATAGGG‑3′和 5′‑AAACCCCTATTCTGTCGAATATCGAGC‑3′(C15)/5′‑ATTGACAGTAGTGGAAAGTATGTCCGG‑3′和5′‑AAACCCGGACATACTTTCCACTACTGT‑3′(C16)。使用Bsa I酶切AtU6‑26‑sgRNA‑SK质粒,将目标基因的靶点序列连入酶切后的AtU6‑26‑sgRNA‑SK质粒,将其转化大肠杆菌感受态细胞Top10,涂含有Amp抗性的LB平板,挑选单克隆利用AtU6‑26‑sgRNA‑SK载体上正向引物SK‑gR‑NA‑F:5′‑CTCACTATAGGGCGAATTGG‑3′和靶点序列反向引物C15NtREV‑T1R:5′‑AAACCCCTATTCT‑GTCGAATATCGAGC‑3′做菌落PCR鉴定并测序。将测序正确的重组子用Spe I和Nhe I进行双酶切,回收酶切片段即sgRNA cassette,同时用Spe I 酶切pCAMBIA1300‑pYAO‑Cas9质粒,将含有靶点序列的sgRNA cassette片段连入pCAMBIA1300‑pYAO‑Cas9载体,将其转化大肠杆菌Top10,涂含有Kan抗性的LB平板,挑选单克隆利用表1中位于Spe I上下游的引物做菌落PCR鉴定并测序。

表1 本研究引物序列及用途Table 1 Sequences and purpose of primers in this study
1.2.2 烟草REV基因转化及阳性苗检测 将构建成功的pCAMBIA1300‑pYAO‑Cas9重组子转化农杆菌感受态细胞 LB4404,在含有抗性的LB固体培养基(50 μg/mL Kan, 25 μg/mL Rif)上进行阳性克隆筛选,并通过载体引物和靶点序列引物检测阳性菌落(表1)。挑取阳性菌落利用农杆菌介导的叶盘转化方法进行烟草遗传转化,并收集T0代种子[21]。取上述收获的T0代烟草种子,用0.1%次氯酸钠和75%酒精进行表面消毒后,利用无菌水清洗干净置于超净台灭菌滤纸上,待风干之后,将种子撒播在1/2 MS固体培养基(25 μg/mL Hyg)培养15 d主根长于2-3 cm的幼苗,移栽到草炭土中放入人工气候箱继续培养(温度25℃、湿度70%、16 h光照/8 h黑暗、4 000 lx)。同等条件下种植野生型烟草作为对照。等幼苗长出5-7片叶后,取叶片,通过CTAB法分别提取待检测阳性苗和对照苗的基因组DNA,根据pCAMBIA1300‑pYAO‑Cas9质粒的Hyg基因设计特异性扩增引物(表1),鉴定烟草阳性苗。
1.2.3 烟草REV突变体的检测 依据靶点序列设计上下游检测引物(表1),利用CTAB方法提取阳性苗基因组DNA,通过PCR扩增含有靶点序列的烟草REV基因DNA序列,同时将烟草野生型材料作为对照。扩增产物通过回收直接送测序公司进行测序。利用Chromas软件和DNAMAN软件对所有测序获得的序列和烟草参考基因组REV基因序列进行比对分析,并统计编辑形式,选取纯合突变体用于后续表型分析。
1.2.4 烟草REV突变体的顶芽、叶片、腋芽表型观测 将上述检测的发生REV基因纯合编辑烟草幼苗,播种草炭土中放入人工气候箱继续培养,生长10 d,用体式显微镜观察观测顶芽生长情况,同时利用解剖镜解剖REV基因编辑纯合突变体的烟草顶端分生组织,参考王兵[22]扫描电镜样品制作方法,通过用2.5%的戊二醛固定烟草解剖组织,0.1 mol/L的磷酸缓冲液多次清洗,再用乙醇浓度梯度逐级进行脱水,将脱水的材料置入乙酸异戊酯置换乙醇,然后将烟草样品放入液态CO2临界点干燥,最后把样品黏在有双面导电胶的金属载台上,放入离子溅射仪中喷金,置于扫描电子显微镜中观察,工作电压为10 kV,观察记录烟草顶端分生组织特点,并选择具有代表性的视野拍照记录,同时将烟草野生型材料作为对照。
2022 年在贵州省烟草科学研究院福泉基地种植正常生长的栽培烟草纯合突变体和对照 K326,采用小区试验,3次重复,每个重复种植 30 株,行距1.1 m,株距 0.55 m,移栽密度 16 500 株/hm2,施氮量105 kg/hm2。参照烟草行业标准《 YCT142‑2010烟草农艺性状调查测量方法》,在盛花期测量每个重复生长相对一致的代表性9株烟草的自然株高和着生叶数,中心花开放50%时进行打顶同时去除已长出的腋芽,在打顶后 28 d,一次性采集烟株的腋芽(打顶后生长的)并称重。
1.2.5 数据分析 试验数据采用SPSS Statistics 17.0软件进行统计分析。采用比较均值(独立样本t检验)分析编辑基因的材料和对照组的自然株高、自然叶数、腋芽鲜重等农艺性状的差异。
2 结果
2.1 sgRNA克隆框的构建
通过数据库,获得烟草REV基因序列,结果表明NtREV基因含有两个拷贝数,即NtREV1(Nitab4.5_0004624g0050.1)和NtREV2(Nitab4.5_0003131g0030.1),全长分别为6 111 bp和6 105 bp,NtREV含18个外显子和17个内含子,NtREV氨基酸序列分析表明从N端到C端含有4个保守结构域,分别是HD结构域、Zip结构域、START结构域和MEKHLA结构域,通过比对烟草NtREV氨基酸序列,NtREV1和NtREV2氨基酸同源性高达98.93%(图1)。为了有效编辑烟草中NtREV,在NtREV基因的第一个外显子上选取“脱靶可能性”较低的2个靶位点,靶点引物列于表1,下划线表示PAM序列(图2‑A)。分别将合成的烟草NtREV单靶点引物经过引物变性退火、Bsa I酶切和T4 DNA连接酶成功构建AtU6‑26‑sgRNA‑SK载体,转化大肠杆菌、挑单菌落、扩菌、提取质粒,用Nhe I和Spe I 对提取的AtU6‑26‑sgRNA‑SK‑NtREV重组子质粒通过双酶切进行核酸电泳,结果显示,泳道1-10为NtREV靶点sgRNA表达盒和NtREV靶点序列,其中sgRNA表达盒序列和NtREV靶标序列大小分别为3 507 bp 和642 bp,泳道11为AtU6‑26‑sgRNA‑SK‑NtREV重组子质粒,表明NtREV靶标序列已成功连入AtU6‑26‑sgRNA‑SK载体(图2‑B)。

图1 NtREV1(Nitab4.5_0004624g0050.1)和NtREV2(Nitab4.5_0003131g0030.1)氨基酸序列的同源性比较Fig. 1 Homology comparison of NtREV1 and NtREV2 amino acid sequences
2.2 植物双元表达载体pCAMBIA1300‑pYAO‑Cas9‑NtREV的构建
为了将AtU6‑26‑sgRNA‑SK‑NtREV重组子中sgRNA表达盒序列连入线性化的 pCAMBIA1300‑pYAO‑Cas9载体,使用Spe I酶切pCAMBIA1300‑pYAO‑Cas9载体,同时将上述获得Nhe I和Spe I酶切回收的产物与Spe I酶切pCAMBIA1300‑pYAO‑Cas9载体通过T4 DNA连接酶连接,转化大肠杆菌感受态细胞,挑取12个单克隆使用载体特异引物1300‑gRNA‑F和1300‑gRNA‑R进行PCR扩增初步鉴定sgRNA表达盒序列是否连入表达载体(表1),实验结果表明,泳道1-11均扩增出sgRNA表达盒条带,泳道12未扩增出sgRNA表达盒条带(图3‑A),最后挑选携带重组子的单菌落扩菌,提取质粒,通过Xba I 和 Kpn I 进行酶切验证pCAMBIA1300‑Pyao‑Cas9‑NtREV最终载体是否构建成功,实验结果表明通过双酶切获得大小750 bp的含有NtREV靶标的表达盒序列(图3‑B)。
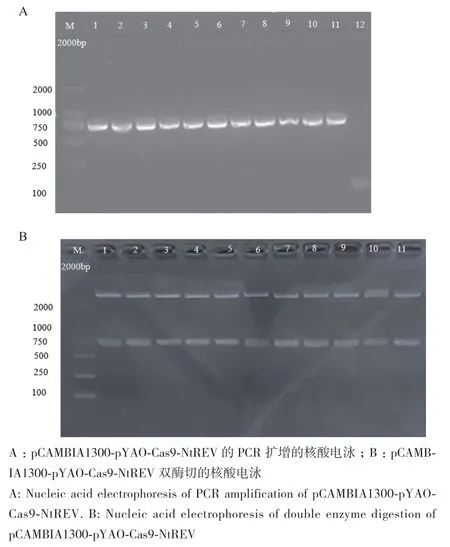
图3 植物双元表达载体pCAMBIA1300-pYAO-Cas9-NtREV的构建Fig. 3 Construction of the plant binary expression vector pCAMBIA1300-pYAO-Cas9-NtREV
2.3 烟草转基因及阳性苗的筛选和检测
分别将两个单靶点的重组子pCAMBIA1300‑pYAO‑Cas9‑NtREV质粒转化农杆菌感受态细胞,通过农杆菌介导的叶盘转化法浸染烟草,将烟草叶片置于含有Hyg抗性的培养基,共获得48株能在培养基中正常生根的幼苗,其中靶点C15植株为24株,靶点C16植株为24株(图4)。为进一步确认抗性苗为转基因植株,提取抗性苗叶片基因组DNA,以Hyg基因特异引物对Hyg‑F和Hyg‑R进行PCR扩增检测(表1),结果(图4)显示,48株待检测材料中有44株材料的DNA模板扩增出约700 bp目的条带,表明成功获得44株REV转基因编辑植株,总体转化效率为90.90%,其中靶点C15植株为21株,C15植株转化效率为87.50%(图4‑A),靶点C16植株为23株,C16植株转化效率为95.83%(图4‑B)。
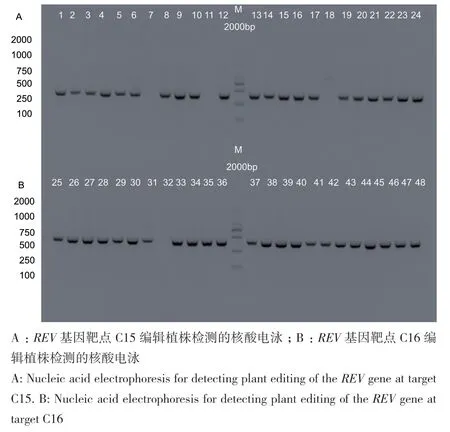
图4 转基因植株检测的核酸电泳Fig. 4 Nucleic acid electrophoresis for the detection of transgenic plants
2.4 烟草NtREV突变体的编辑形式检测
为了验证构建的NtREV基因编辑载体的有效性,用NtREV‑CRISPR test‑F/R引物对上述NtREV基因C15和C16的单靶点编辑T0代植株和野生型基因组DNA进行PCR扩增并通过测序分析编辑形式(表1)。测序结果表明,靶点C16编辑Ko‑C16Ntrev基因突变体4株,其中NtREV基因靶点C16编辑的双基因纯合突变体1株,Ko‑C16Ntrev‑2编辑形成为NtREV1靶点PAM区域上游4位置的A碱基被删除和NtREV2靶点PAM区域上游3位置的A碱基被删除,距离NtREV起始密码子183 bp位置缺失T碱基,导致NtREV氨基酸第60位置之后的移码突变(L61S62→C61R62);杂合编辑突变体3株,Ko‑C16Ntrev‑1编辑形成为NtREV1并未发生编辑和NtREV2靶点PAM区域上游4位置的A碱基被删除,距离NtREV起始密码子181 bp位置A碱基被替换成T碱基,导致NtREV氨基酸第60位置异亮氨酸突变为苯丙氨酸(I60→F60);Ko‑C16Ntrev‑3编辑形成为NtREV1靶点PAM区域上游1位置的A碱基被G替换,上游4位置的A碱基被删除,上游6位置的A碱基被T替换和NtREV2靶点PAM区域上游4位置的A碱基被删除,距离NtREV起始密码子183 bp位置缺失T碱基,180 bp 位置T碱基被替换成C碱基,185 bp位置 T碱基被替换成A碱基,导致NtREV氨基酸第59位置和第62位置未发生突变,NtREV氨基酸第60位置之后的移码突变(L61S62→C61R62);Ko‑C16Ntrev‑4编辑形成为NtREV1并未发生编辑和NtREV2靶点PAM区域上游2位置处的A替换T,距离NtREV起始密码子181 bp位置A碱基被替换成T碱基,导致NtREV氨基酸第60位置异亮氨酸突变为苯丙氨酸(I60→F60)。
靶点C15编辑Ko‑C15Ntrev基因突变体4株,其中NtREV基因靶点C15编辑的双基因纯合突变体1株,Ko‑C15Ntrev‑2编辑形成为靶点PAM区域上游5位置插入A碱基和NtREV1靶点PAM区域上游5位置插入A碱基,插入A碱基距离NtREV起始密码子78 bp位置,导致NtREV氨基酸第26位置之后的移码突变(V26R27→C26P27);杂合编辑突变体3株,Ko‑C15Ntrev‑1编辑形成为NtREV1靶点PAM区域上游5位置插入A碱基和上游14位置G碱基替换A碱基,19位置A碱基替换T碱基;插入A碱基距离NtREV起始密码子78 bp位置,64 bp 位置A碱基被替换成T碱基,69 bp位置 T碱基被替换成C碱基,导致NtREV氨基酸第22位置丝氨酸被替换成半胱氨酸(S22→C22)、NtREV氨基酸第23位置氨基酸未发生变化,NtREV氨基酸第26位置之后的移码突变(V26R27→C26P27);Ko‑C15Ntrev‑3编辑形成为NtREV1靶点PAM区域上游5位置插入A碱基和上游13和14位置TG碱基替换CA碱基;插入A碱基距离NtREV起始密码子78 bp位置,69-70 bp TG碱基被替换成AC碱基,导致NtREV氨基酸第23位置丝氨酸被替换成精氨酸和第24位置甘氨酸被替换成精氨酸(S23G24→R23R23)、NtREV氨基酸第26位置之后的移码突变(V26R27→C26P27);Ko‑C15Ntrev‑4编辑形成为NtREV1靶点PAM区域上游5位置处插入A碱基和上游14位置G碱基替换A碱基,插入A碱基距离NtREV起始密码子78 bp位置,69 bp位置 T碱基被替换成C碱基,NtREV氨基酸第23位置氨基酸未发生变化,NtREV氨基酸第26位置之后的移码突变(V26R27→C26P27)(图5)。
2.5 烟草REV突变体的表型观测
与正常发育的野生型(图6‑I)相比,烟草NtREV单靶点的突变体均发生了不同程度顶芽缺失和叶片畸形(图6),其中双拷贝同源突变株系Ko‑C15Ntrev‑2呈现叶片畸形表型,展示出漏斗形叶、荷叶形叶等形态,表明突变体中REV基因功能受到不同程度的影响(图6‑A‑F),而单拷贝同源突变体Ko‑C15Ntrev‑1、Ko‑C16Ntrev‑3 展示植株正常生长(图6‑G, H)。为了进一步分析NtREV在烟草芽发育中的功能,选择了Ko‑C15Ntrev和Ko‑C16Ntrev编辑形式的rev突变体观测表型(图7),通过光镜观察可知,与野生型顶芽发育相比较(图8‑A, D, G),Ko‑C15Ntrev双拷贝同源突变表现出顶芽缺失,无法正常生长(图8‑C, F, I);Ko‑C16Ntrev单拷贝同源突变体顶芽发育正常,但是顶芽发育迟缓(图8‑B, E,H)。通过大田种植统计烟草自然株高、叶片数量和腋芽鲜重,如图9所示,Ko‑C16Ntrev突变体自然株高较野生型增加3.76%(图9‑A),Ko‑C16Ntrev突变体着生叶片数减少21.47%,达到极显著差异(图9‑B, C),相较于WT(图9‑D),Ko‑C16Ntrev突变体腋芽生长形态正常,但腋芽数量和鲜重减少(图9‑E),Ko‑C16Ntrev突变体腋芽鲜重较野生型显著减少23.41%。

图6 突变体材料和野生型材料的叶形态表型Fig. 6 Leaf morphological phenotypes of mutant and wildtype materials

图7 烟草Ko-C15Ntrev和Ko-C16Ntrev突变体编辑形式Fig. 7 Editing forms of Ko-C15Ntrev and Ko-C16Ntrev mutants in tobacco
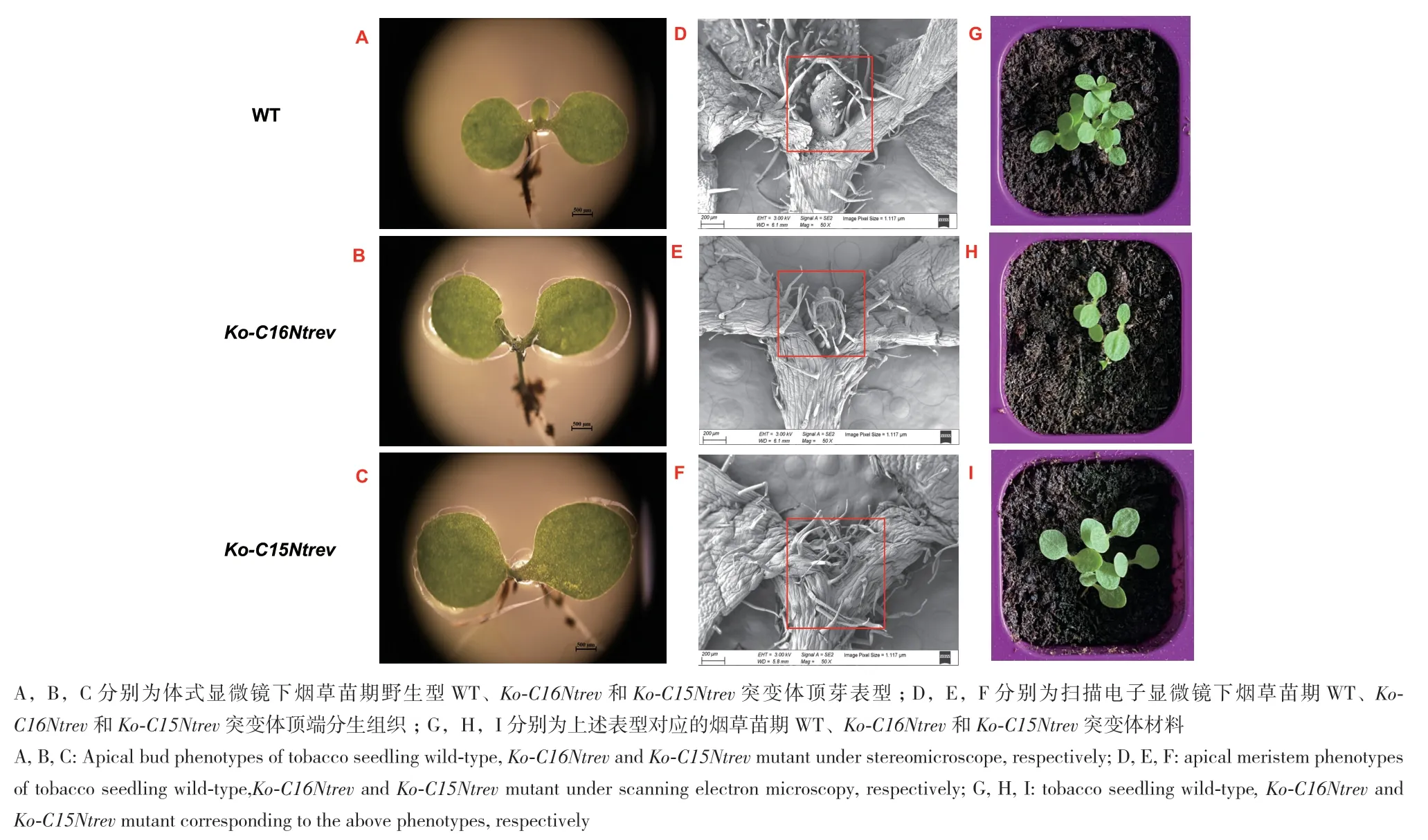
图8 突变体材料和野生型材料的顶芽观测Fig. 8 Observations of apical buds of mutant and wild-type ones

图9 Ko-C16Ntrev突变体的自然株高、叶数、腋芽鲜重表型Fig. 9 Phenotypes of plant height, leaf numbers and fresh weight of C16Ntrev mutants
3 讨论
3.1 烟草NtREV参与调节侧生分生组织的发育
植物侧枝的发育存在精细而又复杂的调控网络,研究报道植物HD‑Zip III转录因子参与调控侧生分生组织的起始,尤其是拟南芥HD‑Zip III基因家族成员中REV(AT5G60690)基因对AM启动的时间和空间精确调控。栽培烟草为异源四倍体,陈浣等[19]研究表明,栽培烟草NtHD‑Zip III基因家族数量是拟南芥的4倍,表明随着染色体加倍,NtHD‑Zip III基因家族成员间功能出现分化以及功能冗余。相比较于拟南芥REV,栽培烟草中REV(NtREV1和NtREV2)存在两个拷贝,因此,研究栽培烟草的REV基因功能需要将两个基因同源拷贝同时敲除才可能实现表型。随着CRISPR/Cas9编辑技术日趋完善,为栽培烟草等多倍体作物的基因功能和突变体创建提供了技术支撑。本研究创制的Ntrev突变体表现出顶芽缺失、叶畸形、叶片数量和腋芽鲜重显著减少,说明NtREV参与调控顶端分生组织、侧生分生组织发育。Shi等[16]证实拟南芥中REV直接调控STM表达量,从而促进侧生分生组织起始。拟南芥ago10突变体研究表明,ARGONAUTE10(AGO10)结合miR165/166,通过降解miRNA,激活REV基因,从而实现调节AM起始[18]。烟草NtREV基因发挥的功能与拟南芥AtREV功能一致,它们均参与了调节侧生分生组织的发育。
烟草NtREV基因存在两个拷贝,不同位置的单靶点的sgRNA表达盒编辑NtREV基因,导致了编辑形式多样化,烟草rev突变体产生了不同的表型,对NtREV基因C16靶点的编辑氨基酸形式进行分析,Ko‑C16Ntrev‑1和Ko‑C16Ntrev‑4材料NtREV氨基酸第60位置异亮氨酸均突变为苯丙氨酸(I60→F60),植株表现出顶芽生长延迟,植株生长正常,而Ko‑C16Ntrev‑2和Ko‑C16Ntrev‑3编辑材料的氨基酸编码的蛋白形式进行分析,结果表明NtREV氨基酸第60位置之后的移码突变(L61S62→C61R62),植株表现出顶芽畸形、叶畸形等性状。以上结果表明REV蛋白HD结构域可能参与调节株型发育相关的基因,尤其是REV蛋白HD结构域第60位置异亮氨酸调节分生组织组织分化,最终影响植株形态建成。这一结果与水稻REV缺失突变体lf1表现出侧生器官极性发育缺陷的表型一致[23]。此外,具有稳定遗传性状的Ko‑C15Ntrev和Ko‑C16Ntrev突变体表型最明显,Ko‑C15Ntrev顶芽缺失,Ko‑C16Ntrev叶片数量和腋芽生物量显著下降,这些突变体还表现出叶畸形、叶漏斗形、叶荷叶形等性状变异,这些表型的出现可能是由于脱靶效应导致假表型,也可能是由于编辑NtREV不同结构域导致功能差异[4,24]。上述突变体为后续通过转录组学建立烟草NtREV调控叶芽发育的分子网络提供了重要研究材料。
3.2 烟草NtREV通过调控生长素合成影响侧芽发育
烟草NtREV结构分析表明存在HD‑Zip III蛋白4个保守结构域,HD和Zip结构域比较保守,参与调控靶基因的表达,而START和MEKHLA结构域在动物研究中较多,参与了外界信号接收与传递[25-27]。然而,植物中关于HD‑Zip III蛋白START和MEKHLA结构域发挥的作用报道较少。目前,在植物中仅知道PYR蛋白START结构域充当ABA的受体结合脱落酸,调控脱落酸的信号通路[28]。后续可利用CRISPR/Cas9编辑技术对NtREV的 START和MEKHLA结构域进行定点编辑,分析其功能。
植株通过IAA极性运输,建立生长素浓度梯度调节植物顶端优势。拟南芥REV缺失突变体ifl1生长素转运蛋白PIN1的定位发生异常,生长素的极性运输能力急剧下降,表现出植物顶端优势丧失。Brandt等[29]发现REV通过直接结合生长素合成途径的2个关键酶TAA1和YUCCA5的启动子调控元件序列(ATG/CAT)参与了生长素的合成。这些结果表明,REV在转录水平上与生长素关系密切。本研究创制的烟草NtREV突变体表现出顶芽缺失、叶片畸形,这可能是由于烟草NtREV中MEKHLA结构域作为受体结合生长素诱导的信号分子改变REV构象,形成二聚体激活下游生长素合成的关键基因引起的表型。本研究证明了REV参与顶端分生组织和腋芽的发育。然而,栽培烟草中REV与生长素合成间的关系,有待下一步利用上述突变体材料开展NtREV调控生长素合成分子机制的研究。
4 结论
本研究通过CRISPR/Cas9编辑系统成功编辑了栽培烟草中的NtREV基因,获得了该基因不同单靶点的突变体,靶点C15双拷贝同源突变体Ko‑C15Ntrev表现出顶芽缺失表型,而靶点C16单拷贝同源突变体Ko‑C16Ntrev表现出烟草叶片数量和腋芽鲜重显著减少,表明NtREV参与了烟草顶端分生组织和腋芽发育,同时也说明基因编辑位点不同将导致植株表型出现差异。
