Schnitzler综合征1例并文献复习
2023-11-22黎展宏唐曼曼黄嘉琪梁云生
黎展宏,唐曼曼,黄嘉琪,梁云生
患者,男,63岁,因“全身红斑风团伴发热、关节痛4年”于2021年1月22日入院。患者4年前无明显诱因躯干四肢出现红斑风团伴瘙痒;风团24 h内不消退,消退后留有褐色色素沉着;期间伴发热(37.5~38.5℃),全身多处关节疼痛。患者症状反复发作,使用抗组胺药症状未见好转,外院予“泼尼松12 mg/d、沙利度胺25 mg/d、雷公滕多苷片40 mg/d”治疗,症状见好转。1个月前复发就诊我院,患者既往史、家族史无特殊。皮肤专科检查:全身淋巴结未扪及肿大;躯干、四肢可见泛发性片状不规则红斑、暗红斑,部分红斑融合成片,局部可见褐色色素沉着,皮肤划痕试验(-)。实验室检查示RBC 3.16×1012/L, WBC 12.59×109/L,NEU 10.15×109/L,ESR 135 mm/h,铁蛋白319.58 μg/L;ALT 108.01 U/L,Alb 34.09 g/L;补体因子C3 1.21 g/L,补体因子C4 0.22 g/L,IgM 24.87 g/L,IgA 0.29 g/L;CRP 32.91 mg/L;细胞因子水平检测:白介素1(IL-1) 168.2 ng/L,IL-22 1 168 ng/L,IL-6<5.1 ng/L,巨噬细胞炎性蛋白51.3 ng/L;血清蛋白电泳:白蛋白 49.73%,α1球蛋白6.79%,γ球蛋白25.37%,M蛋白含量10.26%;血清免疫固定电泳: IgM κ型;基因突变检测:MYD88突变阳性、CXCR4突变阳性。抗核抗体、IgE未见异常。皮肤及皮下组织病理:表皮轻度萎缩变薄,真皮浅层血管内见少许中性粒细胞,血管周围少许淋巴组织细胞、个别中性粒细胞和少许核尘(图1)。根据患者临床表现及实验室检查结果,明确诊断Schnitzler综合征。入院后给予秋水仙碱(25 mg/d)治疗,症状部分缓解。患者治疗2周后出院加用甲泼尼龙(4 mg/d)。患者症状基本控制,目前规律随访中。
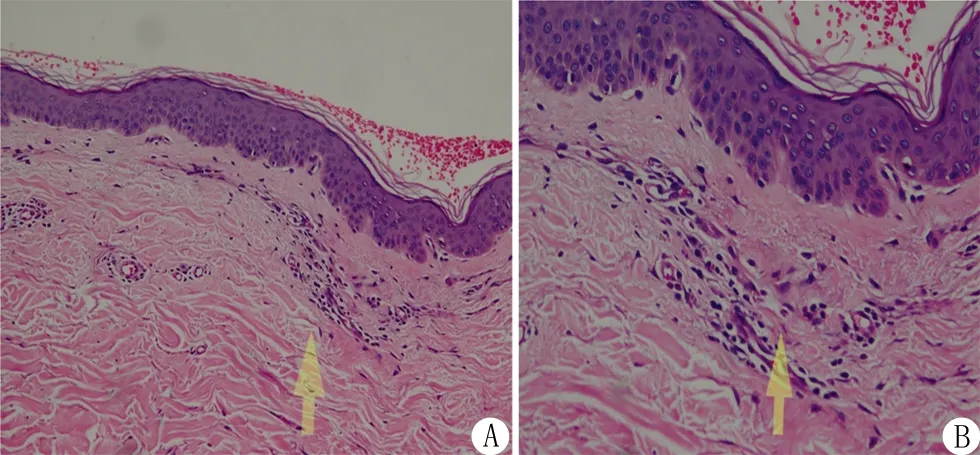
注:真皮浅层血管内见少许中性粒细胞,血管周围少许淋巴组织细胞、个别中性粒细胞和少许核尘(A.×100,B.×200)。
讨 论Schnitzler综合征(Schnitzler syndrome,SS)是一种累及全身的罕见自身免疫性疾病。该病最早由法国皮肤病学专家Schnitzler于1972年发现并描述,目前国内外仅报道了300余例。SS好发于50岁左右的人群,男性发病率略高于女性;目前尚未报道未成年人患病。SS临床表现多样,极其容易被误诊、漏诊。SS标志性的症状为荨麻疹样皮损和单克隆免疫球蛋白(IgM或IgG)上升;病理学可表现为真皮浅层血管周围的中性粒细胞浸润。SS还可出现的临床表现是:ESR升高、发热、关节炎(痛)、白细胞升高、骨痛、骨骼形态异常、淋巴结肿大、瘙痒症、肝脾大、肾损伤等[1]。
SS无特异性的诊断方法。目前常用Strasbourg诊断标准(表1),其主要标准2条:慢性荨麻疹样皮损,IgM或IgG升高;次要标准4条:发热(>38℃),骨受累,病理提示NEU浸润,CRP>30 mg/L和/或WBC >10×109/L。在符合全部主要标准后,若患者IgM升高再满足2条次要标准即可明确诊断。发热、炎性指标异常等在自身炎性疾病中较为普遍[2],但IgM增高有重要的临床意义。当患者出现荨麻疹样皮损伴发关节痛等系统表现时,可考虑从遗传性cryopyrin相关性周期性综合征(CAPS)、成人Still综合征、荨麻疹性血管炎之间鉴别:(1)CAPS:发病年龄在出生时。(2)成人Still综合征:参照Yamaguchi标准,皮疹呈现一过性(伴随发热),实验室检查血清铁蛋白高于正常值5倍以上;病理可见坏死角质形成细胞。(3)荨麻疹性血管炎:可见补体C3、补体C4下降和抗C1q抗体阳性[3]。本患者表现为老年男性、慢性病程。患者主要症状为发热、骨关节痛、荨麻疹样皮损,初步考虑自身炎性疾病;关键检查可见IgM升高、炎性指标升高、病理中性粒细胞浸润。患者满足Strasbourg诊断标准,最终确诊SS。

表1 Strasbourg诊断标准[1]
临床发现IL-1拮抗剂可较好控制SS症状,这提示IL家族在SS的发病中起着重要作用。其可能机制是:外源性的刺激因子介导下游的MYD88调节因子致NF-κB信号通路异常激活,使NLRP3炎性小体表达增加。NLRP3炎性小体可促进IL-1β和IL-18的生成[4]。IL-1β可趋化中性粒细胞,介导中性粒细胞胞外诱捕网的形成[5]。部分研究显示MYD88基因、NLRP3 L256P突变可能与SS发病有关。这一定程度上解释了SS的体征为何与遗传性cryopyrin相关性周期性综合征(NLRP3基因突变)相似,也说明了为何20%未经治疗的SS患者会进展为华氏巨球蛋白症(MYD88基因突变)[6]。本患者MYD88基因突变阳性、IL-1升高,此结果为进一步确诊SS提供依据。
SS目前并无根治手段。治疗上非甾体类抗炎药可减轻关节疼痛。免疫抑制剂疗效确切且经济,如秋水仙碱能够抑制中性粒细胞的趋化。生物制剂靶点精准但昂贵:IL-1受体阻滞剂(卡那单抗等)能够安全有效地改善患者病情;IL-6受体拮抗剂(托珠单抗等)、布鲁顿酪氨酸激酶抑制剂(伊布替尼)或可治疗SS。此外针对NLRP炎性小体的药物已进入2期临床试验[7]。该患者使用秋水仙碱后症状部分控制,加用甲泼尼龙后症状完全控制。目前无专门的量表评估SS病情。皮肤表现直观且易于观察,本例使用慢性荨麻疹量表评估病情。患者“7日荨麻疹活动评分”从28分降至为1分,“荨麻疹控制程度测试”现为13分。
SS患者可进展为血液系统疾病,可会出现致死性的结局[8]。患者外院血液科确诊华氏巨球蛋白血症,目前未达到化疗指征,现密切随访观察中。
