基于CRISPR-Cas9技术构建TET2基因敲除的鸡胚成纤维细胞系
2023-11-22王强州潘诗雨方梦雅李微王佳兴刘茵茵陈世豪
王强州, 潘诗雨, 方梦雅, 李微, 王佳兴, 刘茵茵, 陈世豪*
(1.扬州大学表观遗传学与表观基因组学研究所,江苏 扬州 225009; 2.扬州大学兽医学院,江苏 扬州 225009; 3.江苏省家禽科学研究所,江苏 扬州 225125)
甲基胞嘧啶双加氧酶2 (tet methylcytosine dioxygenase 2,TET2)是双加氧酶家族的成员之一,作为细胞中重要的DNA 去甲基化酶,催化5-甲基胞嘧啶(5mC)转化为5-羟甲基胞嘧啶(5hmC),在细胞基因组表观遗传学中起着重要调控作用[1-2]。近年来,TET2在免疫应答和炎症调控中的作用逐渐被认识,成为免疫学研究热点之一。TET2 对于T 细胞和B 细胞免疫稳态的维持以及相关免疫因子的激活具有重要的调控作用[3-5]。TET2作为启动天然免疫应答的关键分子,在浆细胞样树突状细胞中被CXXC5 招募至TLR7/9 启动子并促进转录,启动一系列免疫级联反应,激活天然免疫应答[6]。小鼠骨髓来源树突状细胞中敲除TET2 蛋白,其Toll-like 受体信号传递受阻[7],表明TET2与Toll-like受体信号通路密切相关。本团队前期研究证实,DNA 甲基化参与调控鸡的抗病毒天然免疫反应[8],而鸡TET2 作为去甲基化过程的关键蛋白,对天然免疫的调节作用很大程度上未知。鸡胚成纤维细胞系DF-1 是自发永生化形成的细胞系[9],是研究鸡天然免疫调控机制的理想细胞系。因此,构建稳定的TET2敲除的鸡胚成纤维细胞系,可为进一步研究鸡TET2在天然免疫反应中的调控作用提供可靠的体外细胞模型。
CRISPR-Cas9 是新一代基因编辑技术,通过人工设计的向导sgRNA,引导Cas9 核酸酶在基因组特定位点进行靶向编辑,包括基因敲除(knockout)、敲入(knockin)[10-11]。该技术已广泛应用于细胞敲除、基因筛选、基因治疗、分子育种等领域,尤其在构建敲除细胞或敲除动物模型方面具有不可替代的作用,也为动植物的分子育种提供了新的技术与思路[12-13]。本研究利用CRISPRCas9 技术构建敲除TET2基因的鸡胚成纤维细胞系,通过靶向干预TET2基因的表达,为进一步探索鸡TET2在天然免疫激活和消退中的功能及分子机制奠定基础。
1 材料与方法
1.1 试验材料
1.1.1细胞和质粒 鸡胚成纤维细胞系DF-1 源于美国ATCC,由本课题组传代保存。非病毒载体Cas9-T2A-GFP 由本课题组通过MLM3613 Cas9质粒(Addgene,#42251)改造而来,携带绿色荧光蛋白(green fluorescent protein,GFP) 标签[11]。pEX-A-U6-sgRNA购自Addgene(#65626)。
1.1.2试剂 DMEM 高糖培养基、胎牛血清购自美国Gibco 公司;DL2000 DNA Marker、2×Phanta Max Master Mix (Dye Plus)、FastPure Gel DNA Extraction Mini Kit、FastPure Cell/Tissue DNA Isolation Mini Kit 购自诺唯赞生物科技股份有限公司(南京);DH5α 感受态细胞,ExpressCast PAGE 彩色凝胶快速试剂盒和化学发光试剂盒均购自新赛美生物科技有限公司;Triton X-100 和Proteinase K(20 mg·mL-1)购自碧云天生物技术有限公司;亚甲基蓝染色液(0.2%)购自北京索莱宝科技有限公司,用ddH2O 稀释至0.02%;无内毒素质粒提取试剂盒Endo Free Plasmid Midi Kit 购自OMEGA 公司;BbsI限制性内切酶、T7核酸内切酶I (T7E I)购自NEB 公司;T4 连接酶购自Thermo Scientific 公司;HighGene 转染试剂购自ABclonal公司;尼龙膜(正电荷)购自Whatman 生物公司;兔源TET2 多克隆抗体购自CST 公司;兔源Actin多克隆抗体购自ABclonal公司;兔源5hmC多克隆抗体购自Active Motif 生物公司;HRP 标记亲和纯化羊抗兔IgG购自Abcam公司。
1.2 试验方法
1.2.1靶向鸡TET2 基因的sgRNA 设计 通过NCBI 在线数据库检索鸡TET2基因组序列,NCBI登录号为NM_001277794.3,其另一个剪切变体登录号为XM_015276223.3。在2 个mRNA 的共有序列设计sgRNA,即把NM_001277794.3序列的第2 个外显子,XM_015276223.3 序列的第3 个外显子作为靶标设计区域。利用sgRNA 在线设计工具(https://chopchop.cbu.uib.no/),按照CRISPRCas9靶点设计原则,设计4对针对TET2CDS 序列的sgRNA,且预测其在基因组内无其他靶点。在靶序列正义链的5’端前部添加caccg,同时合成sgRNA互补的寡核苷酸,并在5’端添加aaac,3’端添加c,合成的正反向oligo DNA序列见表1。

表1 sgRNA寡核苷酸序列Table 1 Sequences of sgRNA oligonucleotide
同时依据目的基因的序列,利用NCBI引物设计工具设计CRISPR-Cas9 敲除鉴定引物,并由苏州金唯智生物科技公司合成。鉴定引物序列如下:TET2-ck1-F:CAATCTGCTATCCTGAGTGA和TET2-ck1-R:ATTGGAGATGCTTGGTGTT,用于检测sgRNA-1 和sgRNA-2 位点;TET2-ck2-F:GCAGCACCCAGAAGAAAT和TET2-ck2-R:AGCAGGGAATCCATCTTTG 用于检测sgRNA-3和sgRNA-4位点。
1.2.2靶向鸡TET2 的sgRNA 载体的构建 将合成的4 对反向互补的sgRNA oligo DNA 单链退火,形成带有粘性末端的短双链DNA。退火步骤如下,将正反向sgRNA 序列以100 mmol·L-1溶于ddH2O,配制以下体系:1 µL Forward oligo,1 µL Reverse oligo,2 µL 10× NEB buffer 2,16 µL ddH2O,置于100 ℃水,自然降温至25 ℃左右即可。
将短双链DNA 与经BbsI 酶切后的质粒pEXA-U6-sgRNA 连接,并将连接产物转化至DH5α 感受态细胞,涂菌板后置于37 ℃过夜培养。挑取单克隆菌落,送苏州金唯智生物科技公司测序鉴定,显示4 对sgRNA 序列均正确插入sgRNA 表达质粒,提示载体构建成功,提取质粒备用。
1.2.3sgRNA 切割效率的测定 DF-1 细胞铺于6 孔板,待生长至80%密度,每个孔分别转染4 µg的sgRNA 和Cas9-T2A-GFP 2 µg,不转染的细胞作为对照,转染6 h 后换成含10% FBS 的DMEM 高糖完全培养基,48 h 后收获细胞,利用FastPure Cell/Tissue DNA Isolation Mini Kit 提取基因组DNA。
分别以TET2-ck1 和TET2-ck2 上下游引物,PCR扩增sgRNA-1、sgRNA-2和sgRNA-3、sgRNA-4靶位点附近的DNA 片段。PCR 反应体系为50 µL:5× SF Buffer 10 µL,Phanta Super-Fidelity DNA Polymerase 1 µL,dNTP Mix (10 mmol·L-1each) 1 µL,上、下游引物各2 µL,模板DNA 2 µL,无酶水32 µL。PCR 扩增条件:95 ℃预变性30 s;95 ℃变性10 s,58 ℃退火10 s,72 ℃延伸15 s,30 个循环;72 ℃充分延伸5 min 后,进行退火。退火条件:95 ℃变性5 min,以0.1 ℃·s-1的速率降温至25 ℃。将退火后的PCR 产物进行纯化,用T7E1 酶37 ℃酶切2 h,其产物用2 %琼脂糖凝胶电泳,分析不同位点的sgRNA 引导Cas9 蛋白的切割效率。
1.2.4微量DNA 提取 待96 孔细胞长至80%~90%融合度,用胰酶消化并重悬细胞,一半继续传代培养,另一半用于DNA 提取,鉴定4 条sgRNA的切割效率。配制微量DNA 提取液:0.01 mol·L-1Tris pH 8,0.002 mol·L-1EDTA,0.2% Triton X-100,200 µg·mL-1蛋白酶K。
2 500 r·min-1离心收集微量细胞(>1 000个),加入50 µL 微量DNA 提取液,充分涡旋后置于56 ℃孵育3 h。孵育结束后,将样本煮沸10 min用于灭活蛋白酶K。短暂离心,将样本收集至管底,冻存至-20 ℃。在50 µL PCR 体系中,取10 µL 作为DNA模板。
1.2.5单克隆敲除细胞株筛选 选用Cas9 蛋白切割活性较高的靶点的sgRNA,用于制备TET2敲除的鸡胚成纤维细胞系DF-1。将DF-1 细胞铺于6 cm 细胞培养皿,培养24 h 至细胞生长至70%融合度,按说明书进行转染。在灭菌EP 管中加入sgRNA 质粒4 µg 和Cas9-T2A-GFP 质粒2 µg,与转染试剂混合均匀,室温孵育10 min。将混合液逐滴加至培养皿中,37 ℃培养6 h。弃去培养基换成含10% FBS 的DMEM 高糖完全培养基,继续培养48 h。用胰酶消化转染48 h 后的DF-1 细胞,制备细胞悬液进行流式细胞分选,以获得高表达GFP 基因的细胞。将获得的GFP 阳性细胞通过有限稀释法,在96 孔板中培养单细胞克隆,待亚克隆的细胞密度生长至80%时,将一半细胞用于提取DNA,另一半细胞继续扩大培养。采用微量DNA 提取法,通过PCR 扩增靶位点序列,送至苏州金唯智生物有限公司进行DNA 测序鉴定。
1.2.6Western blot 分析蛋白水平 将鸡TET2敲除的DF-1细胞系与野生型DF-1细胞系分别铺至6孔细胞板,待细胞密度长至90%左右时收获细胞,裂解获得蛋白,进行Western blot 分析。简要步骤如下:用预冷PBS洗涤细胞沉淀,加入RIPA裂解液进行细胞裂解,冰上孵育30 min;4 ℃,12 000 r·min-1离心15 min。吸上清,通过BCA法测定蛋白含量。然后加入5×SDS-PAGE蛋白上样缓冲液,99 ℃金属浴加热5 min,充分变性蛋白后,进行SDS-PAGE电泳。230 mA 恒流条件下转膜2.5 h。将膜用5%脱脂牛奶封闭缓冲液室温封闭30 min。稀释TET2(1∶1 000)和ACTIN (1∶2 000) 抗体,4 ℃孵育过夜。第2天,洗膜3 次后,用HRP 标记亲和纯化羊抗兔IgG(1∶10 000)室温孵育1 h 后,用TBST 缓冲液洗涤3 次,每次5 min。最后置于蛋白印迹成像系统下观察条带。
1.2.7Dot blot分析方法 将鸡TET2敲除的DF-1细胞系与野生型DF-1 细胞系分别铺至6 孔细胞板,待细胞密度长至90 %左右时,收获细胞,提取基因组DNA,进行Dot blot 分析。基因组DNA 提取方法按照诺维赞FastPure Cell/Tissue DNA Isolation Mini Kit说明书进行。
将DNA 梯度稀释到200、100、50 µg·µL-1,99 ℃变性5 min后置于冰上迅速冷却,放置10 min。然后,将不同含量水平的DNA各上样2 µL到尼龙膜上,室温干燥15 min后,置于80 ℃烘箱加热固定1 h。将固定后的尼龙膜用5 %脱脂牛奶封闭缓冲液室温封闭30 min。稀释5 hmC(1∶1 000)抗体,置于4 ℃孵育过夜。第2 天,用TBST 缓冲液洗膜3 次后,室温下用HRP 标记亲和纯化羊抗兔IgG(1∶10 000)室温孵育1 h,再用TBST洗涤3次,每次5 min。最后置于蛋白印迹成像系统拍照。拍照完成后,尼龙膜用TBST 缓冲液洗涤2 次后,置于0.02%亚甲基蓝染色液染色K,用水漂洗后通过普通相机进行拍照,获得DNA上样内参。
2 结果与分析
2.1 靶向鸡TET2的sgRNA切割效率的测定
琼脂糖凝胶电泳观察目的基因片段被T7E1酶切割情况(图1),结果可知sgRNA-1、sgRNA-2、sgRNA-3 出现被T7E1 酶切割的小片段,说明转染这些sgRNA 后细胞基因组DNA 发生了编辑事件。其中,设计的sgRNA-2 引导Cas9 蛋白对靶标序列表现出较高切割活性,经T7E1酶切后的全长扩增片段(520 bp)产生329和191 bp的片段,大小符合预期。由此,后续筛选单克隆细胞选取sgRNA-2进行转染。
2.2 筛选单克隆敲除细胞株和测序鉴定结果分析
将筛选的单克隆细胞株提取基因组DNA,经PCR 扩增后进行Sanger 测序,测序结果与原始基因组序列进行比对,确定具体突变位置及其突变序列(图2),发现有1 株细胞(命名为DT-6)发生了碱基的缺失突变。TET2-sgRNA-2 切割位点如图2 所示,DNA 双链分别发生了2 和28 bp 的缺失,导致鸡TET2基因移码突变,转录提前终止,推测该DT-6细胞中TET2蛋白将失活。
2.3 TET2敲除细胞形态学变化分析
由图3 可知,与正常DF-1 细胞相比,TET2基因敲除的DT-6 细胞在形态上呈现较为明显的差别,表现为形态多样,较为饱满,呈现无规则的菱形或双锥形,原因可能是TET2敲除导致某些骨架蛋白或黏附蛋白等的表达受到影响。
2.4 敲除细胞TET2 蛋白水平分析
敲除细胞扩大培养后,进行Western blot 验证。结果如图4 所示,相对于DF-1 细胞,敲除细胞系DT-6 中TET2 蛋白几乎不表达,说明敲除TET2 的DF-1 细胞系构建成功。
2.5 敲除细胞羟甲基化水平分析
TET2 维持细胞的羟甲基化(5hmC)水平,可以通过调控基因的羟甲基化水平控制相关基因的表达。本研究利用Dot blot 分析稳定敲除细胞的羟甲基化水平,结果如图5 所示,相对于正常DF-1 细胞,敲除细胞系DT-6 中5hmC 水平显著下降。由此可见,TET2 是维持DF-1 细胞全基因组5hmC 水平的关键蛋白。
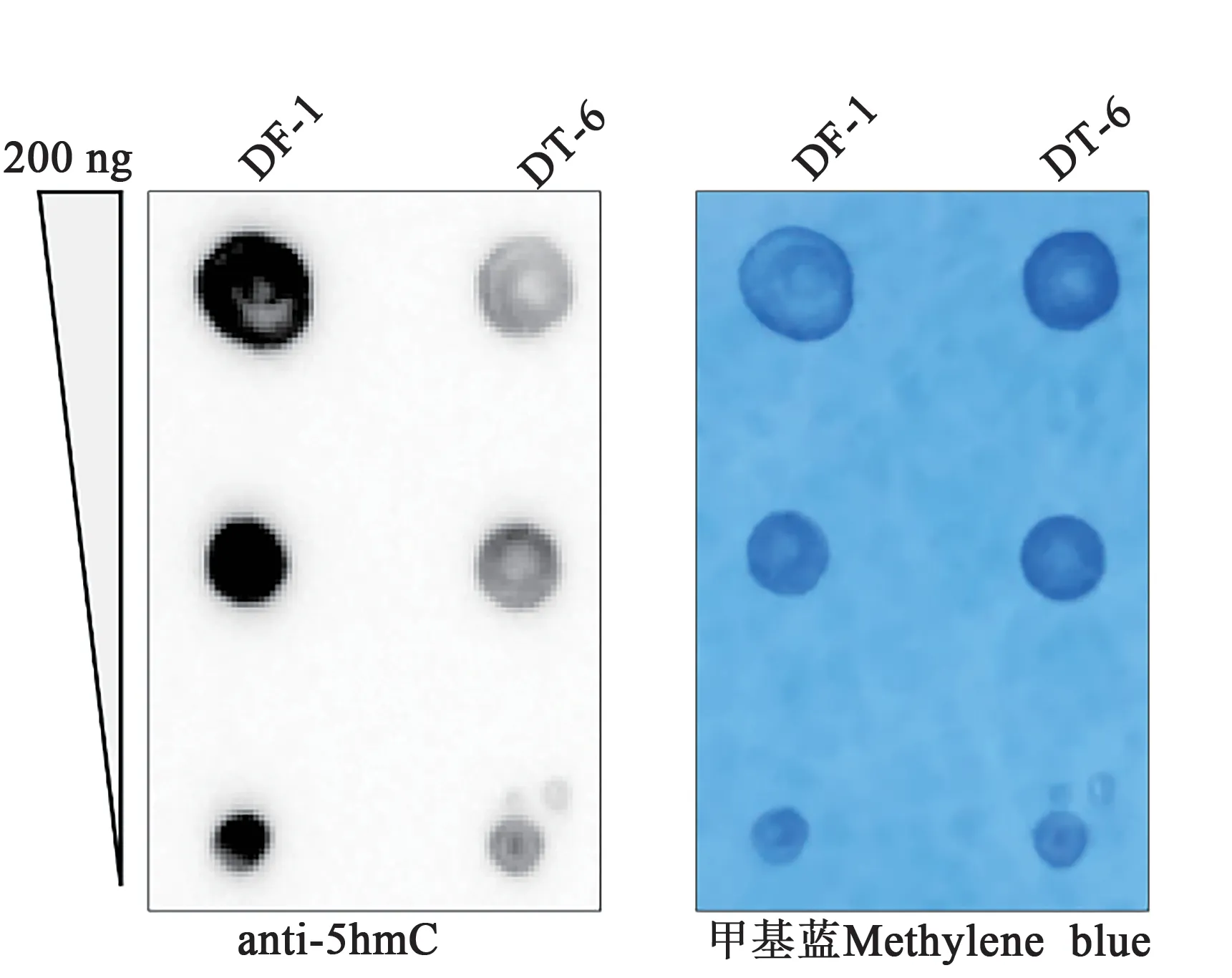
图5 Dot blot检测野生型和TET2敲除细胞的羟甲基化水平Fig. 5 Dot blot detected levels of hydroxymethylation(5hmC) in wild type and TET2 knockout cells
3 讨论
已有研究表明,DNA甲基化抑制剂显著增强鸡胚成纤维细胞的抗病毒天然免疫反应[9],表明DNA甲基化修饰参与调控鸡天然免疫反应的激活。而羟甲基化酶TET2 是一种内源性的DNA 去甲基化酶,对于调控DNA甲基化的平衡具有重要作用[14-15]。但TET2基因及其介导的羟甲基化修饰在鸡抗病毒天然免疫反应中的功能及其作用机制尚未明确。
CRISPR-Cas9 是新一代基因编辑技术,通过人工设计的sgRNA 来识别目的基因组序列,并引导Cas9 蛋白酶在原间隔序列临近基序(protospacer adjacent motif, PAM)区上游的3~8 碱基处切割目的DNA,形成双链断裂,从而启动细胞内的DNA 损伤修复机制,造成基因敲除或敲入,最终达到对基因组DNA 进行编辑的目的[16-17]。与上一代基因编辑技术,如锌指核酸酶(zincfinger nucleases,ZFN)和类转录激活因子效应分子核酸酶(transcriptionactivation-like effector nucleases,TALEN)相比,CRISPR-Cas9 基因编辑技术因操作简单、编辑效率高、试验周期短等特点被广泛用于构建各种细胞系和各种用途的动物疾病模型[17-18]。应用CRISPR-Cas9 技术构建基因敲除的细胞也是禽类基因功能研究的重要手段之一。例如通过构建鸡TBK1 敲除的DF-1 细胞,分析了鸡TBK1在抗病毒信号通路中的功能[19]。
鸡TET2基因位于4号染色体,由11个外显子和10 个内含子组成,该基因也存在另一种剪接变体,由13个外显子和12个内含子组成。sgRNA的设计普遍共识是选择靶基因的第1、第2外显子区域,为保证能够将鸡TET2蛋白彻底敲除失活,本研究选择在鸡TET2 基因的CDS 区的共有序列进行sgRNA 靶点设计,与预期结果一致,该位点sgRNA可以完全敲除TET2基因。本研究还发现,TET2敲除后鸡成纤维细胞发生了明显的形态变化。研究表明,TET2调控诸多发育相关的基因[20-21],鸡TET2还通过DNA去甲基化作用方式调节成肌调节因子的表达,促进鸡成肌细胞的分化[22]。本研究中,TET2敲除是否影响鸡胚成纤维细胞骨架蛋白等基因表达造成细胞形态的变化,还有待进一步验证。
已有研究指出,TET2 对于T 细胞和B 细胞免疫稳态的维持以及相关免疫因子的激活具有重要的调控作用[3-6],也是树突状细胞启动天然免疫应答的关键分子,且与Toll-like 受体信号通路密切相关[7]。上述研究表明,TET2 与免疫细胞功能密切相关,且依赖其DNA 去甲基化的方式来发挥免疫调控作用。本研究利用CRISPR-Cas9技术敲除TET2基因后,DF-1 细胞全基因组5hmC 水平显著下调,表明鸡TET2对于维持细胞基因组羟甲基化水平具有重要作用。鉴于TET2 对免疫反应的激活具有重要的作用,推断TET2敲除后将显著影响鸡胚成纤维细胞天然免疫反应的激活。
综上所述,本研究在未引入抗性筛选基因的条件下获得了TET2基因敲除的单克隆细胞株,不必考虑抗性基因的插入对目标基因功能的影响,为其他类型细胞的筛选提供了适合的方法。本研究应用CRISPR- Cas9 技术成功筛选获得TET2基因敲除的鸡胚成纤维细胞系(DT-6),为研究鸡TET2的生物学功能提供可靠的体外模型,同时为在其他鸡细胞系TET2的敲除提供了有效靶点。
